Development of a Display and Retrieval System for the Protein Data Bank on Sun Microsystems Workstation
Takeshi UNO, Yasuyuki KAWASHIMA, Jinbei ZHANG, Haruhisa HAYASHI,
Kazushige YAMANA, and Hidehiko NAKANO
 Return
Return
INTRODUCTION
In our laboratory, we have developed the molecular display system for small molecules on personal computer named "Modrast-E"[1], and converted it to the workstation[2], for PDB named "Modrast-P" on NEC personal computer[3]. Molecular graphics programs have been reported for some of the common graphical workstations, such as IRIS or TITAN[4,5,6]. However, for the popular Sun SPARC workstation, only a few programs are available for PDB. RASMOL[7] is known as a program which runs on Sun SPARC workstation, but it doesn't have the function to display molecular models clipped by single plane, double plane, or cylinder defined arbitrarily, that Modrast-P has.
In this article, we describe the development of a new molecular graphics system for the Sun SPARC workstation, named "Modrast-P", based on "Modrast-E"[2] and "Modrast-P"[3]. The new features of this system are multi-windows ( text and graphics window ) on the display and user friendly graphical user interface (GUI). Therefore, it is easy for a user to operate by a mouse or buttons. An information retrieval system of the PDB file in the CD-ROM was also developed.
HARDWARE AND SOFTWARE
Modrast-P was developed on Sun Microsystems SPARC series under the operating system of Solaris Version 2.1 or 2.3. This program was written in C language using the library of Xlib for graphics and Xview Version 2.0 for user interface.
DISPLAY SYSTEM OF MOLECULAR STRUCTURE
This system consists of the following functional modules :
A. Coordinate data input / output
B. Display of the molecular structure with various styles
C. Clipping of a molecule by single or double planes
D. Image rotation
E. Specification of a residue in the molecules and of an atom in the
residues
Coordinate data input / output
The input data to this system are PDB[8] source data files which are distributed with the CD-ROM and the Internet. In this system, when a user selects an ID code of PDB, the corresponding PDB file is read from the CD-ROM and so on, and the structures of these molecular models are displayed in one of the graphics windows. The contents of this file can also be shown in text windows. The graphic window can be saved to an image file with popular formats, such as bitmap format, GIF87.
Display styles of the molecular model
This program has four fundamental display styles :
A. Space-filling model
B. Ball-and-stick model
C. Skeleton model
D. Tubular model
In both space-filling and ball-and-stick display styles, there are three coloring modes: by each atom, residue, and chain. In the coloring mode by atom, different elements are painted with different colors. In the coloring mode by residue, different groups of residue are assigned different colors. In the coloring mode by chain, polypeptide chains (protein) or polynucleotide chains (nucleic acid) are assigned different colors. Figure 1 shows the insulin dimer (4INS) drawn with space-filling model. This is drawn in coloring mode by residue.
In the skeleton and tubular display style, molecules are shown with the backbone model with lines or tubes, respectively. With the skeleton model, molecules are drawn in one color with monotonous depth, and therefore can be displayed quickly. The tubular molecular models are shaded images. This model has two coloring modes. One mode is colored by each chain, another by each secondary structure, such as helix, sheet, and turn. Figure 2 shows the insulin dimer (4INS) drawn with tubular model and its coloring mode is by chain. The secondary structure of an alpha helix, which is not easily recognized with space-filling and ball-and-stick models, is clearly observed in this image. The disulfide bonds, which play an important role in keeping a three-dimensional structure, can be shown. In Figure 3, a synthetic DNA (1BNA) is shown with tubular model. Base pairs also can be displayed with tubes, which are shown in this figure.
The tubular model can be displayed together with either a space-filling model or a ball-and-stick model. This model consists of a whole structure with a tube and specified parts with space-filling models or ball-and-stick models. In Figure 4, ferredoxin (1FDX) is shown. This figure shows that there are two inorganic clusters, consisting of iron and sulfur and four cysteine residues connected to one of the clusters as ball-and-stick models. Figure 5 shows the catabolite gene activator protein complex with DNA (1CGP). In this figure, DNA is displayed as a tubular model, and a protein as a space-filling model.
The system has a function to extract and enlarge a part of a compound. In Figure 6, the cluster and cysteine connected to the cluster, extracted from a ferredoxin (1FDX) are displayed as ball-and-stick model.
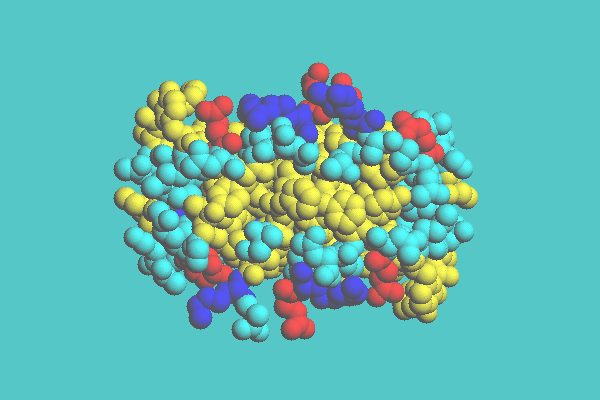
Figure 1. Insulin dimer ( 4INS ) with space-filling model colored by residue.
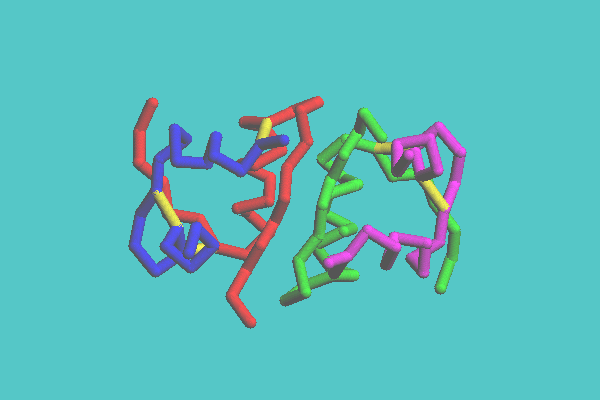
Figure 2. Insulin dimer ( 4INS ) with tubular model colored by chain.
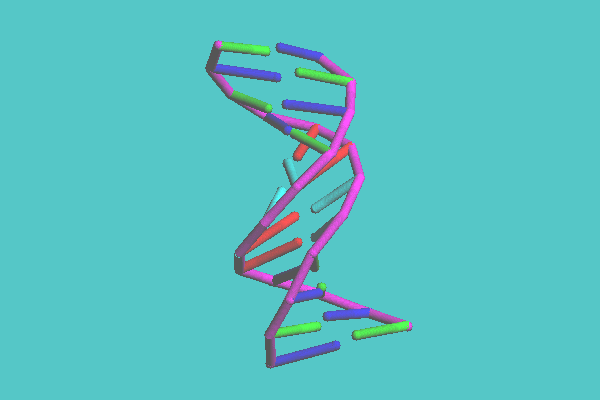
Figure 3. Synthetic DNA ( 1BNA ) with tubular model. Base pairs also can be displayed with tubes.
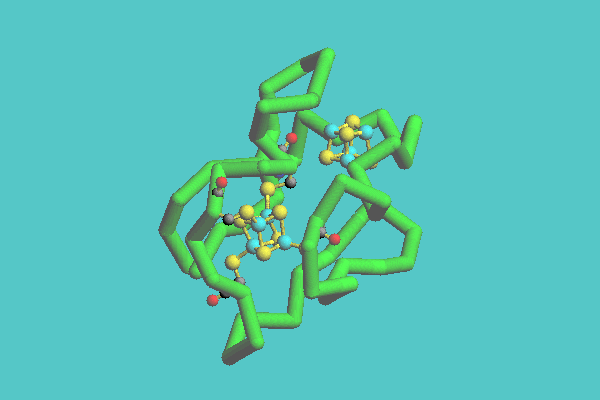
Figure 4. Ferredoxin ( 1FDX ) with heteroatoms displayed as tubular model. In this model, there are two inorganic clusters, composed of iron and sulfur, and four cysteine residues connected to one of the clusters as ball-and-stick models.
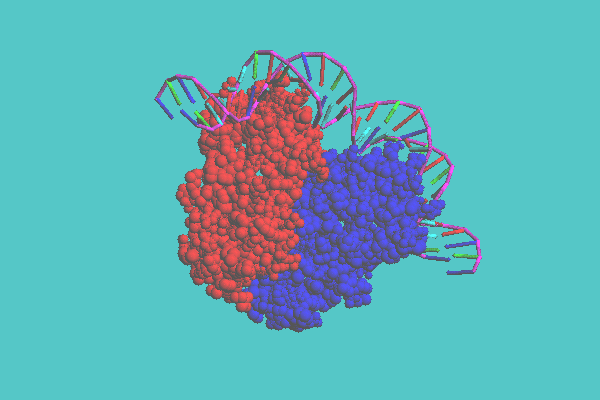
Figure 5. Mixed representation of catabolite gene activator protein complex with DNA ( 1CGP ). In this model, DNA is displayed as a tubular model, and a protein is displayed as a space-filling model colored by chain.
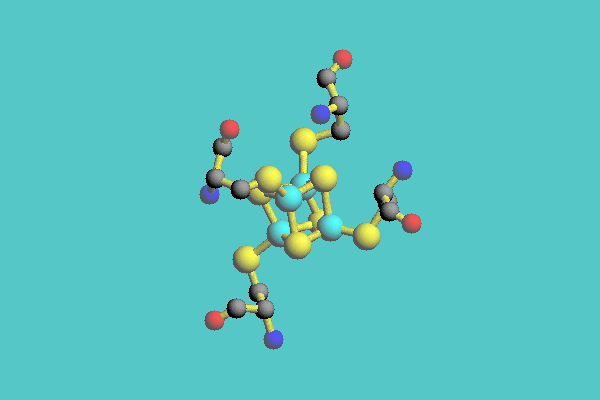
Figure 6. Ball-and-stick model of cluster composed of iron and sulfur, and four cysteine residues connected to the cluster, extracted from ferredoxin ( 1FDX ).
Clipping of a molecule by single or double plane ( s )
In this system, it is possible to display a clipped molecule cut by one or two planes. Using this cutting function, we can observe the inner layers of a displayed molecule as well as the outer shape of a molecule. Methods and display styles are as follows :
A. Space-filling model clipped by one plane
B. Space-filling model clipped by two planes
C. Partially clipped space-filling model, specified part of which is left
D. Clipped space-filling model combined with tubular model
E. Space-filling model clipped by cylinder
The definition of cutting plane(s) was described fully in a previous paper[2]. The first plane is defined by three atoms of one compound. The second plane is to be defined in three different ways : independent of, perpendicular, or parallel to the first plane. In display style E, the cylinder is defined by the radius of it, the direction specified by a vector, and an atom which is located at the center of the bottom of it.
An example of a space-filling model clipped by one plane is shown in Figure 7. Myoglobin is clipped by one plane defined by three atoms on the heme ring. Figure 8 shows an example of space-filling model clipped by two planes, in which myoglobin is cut by two planes, one being defined by the heme ring and the other being perpendicular to the first plane. In Figure 9, ferredoxin (1FDX) is cut by one plane, and a tubular model appears where the space-filling model was cleared off. Figure 10 shows an example of a space-filling model clipped by cylinder. Ferredoxin (1FDX) is cut by a cylinder, whose radius is 75 angstroms. In this representation, one of the clusters of iron and sulfur atoms is specified to be left.
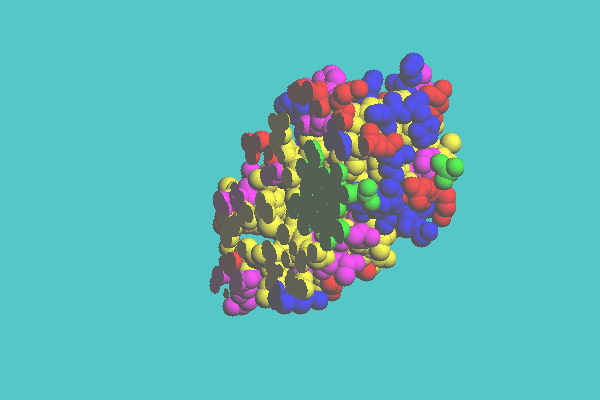
Figure 7. Space-filling model of myoglobin ( 1MBD ) clipped by one plane defined by three atoms on the heme ring. The shape of the heme ring is seen at the cross section.

Figure 8. Space-filling model of myoglobin ( 1MBD ) clipped by two planes. The first plane is the one defined by the heme ring (figure 7), and the second is perpendicular to the first plane.
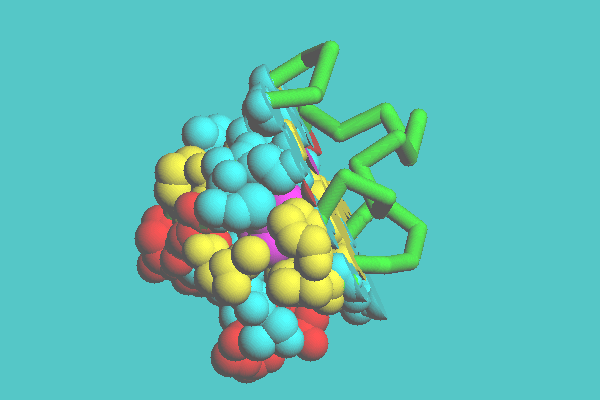
Figure 9. Combined representation of ferredoxin ( 1FDX ). Ferredoxin is cut by one plane, and a tubular model appears where the space-filing model was cleared off.

Figure 10. Space-filling model of ferredoxin ( 1FDX ) cut by a cylinder, the radius of which is 75 angstroms. In this representation, one of the clusters of iron and sulfur atoms, is specified to be left.
Image rotation
By using this function, it is possible to rotate a molecule in the display. The rotation axis can be specified as one of the following three kinds of axes :
A. Coordination axes ( X, Y, Z )
B. Line defined by two molecules
C. Normal of the plane defined by three atoms
One needs to select 2 or 3 atoms for defining the rotation axis except when the coordination axis is chosen. When the rotation axis is determined, a user can rotate the molecule shown with skeleton model in real-time by dragging the slider bar on the display.
Specification of a residue in the molecule and an atom in the residue
The operations to specify an amino acid residue in the molecule or an atom in the residue are numerous. Therefore, we developed this routine as two different methods.
One method is to specify chain, residue name, or sequence number. In this approach, a user can select the target atom by inputting items to the text or selecting in the list items in the panel, as shown in Figure 11. This method is useful for the person who has some knowledge of a protein, a nucleic acid, and so on.
The other method is by pointing the displayed molecular model directly with the mouse. In this approach, only the position of the alpha carbon is specified, but other atoms can be specified in the subwindow in which the structure of the specified amino acid is displayed with wireframe model. A user can employ these two different methods together. For example, if a user needs to specify two atoms, it is possible to use the former to specify the first atom and the latter to specify the second.
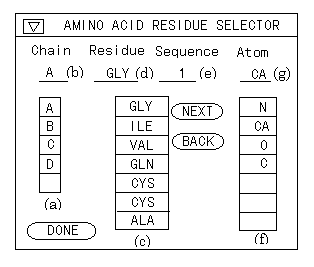
Figure 11 . Amino acid selector. (a) The list of chains. (b) The selected chain. (c) The list of amino acids. (d) The name of the selected amino acid. (e) The sequence number of the selected amino acid. (f) The list of atoms. (g) The name of the selected atom.
INFORMATION RETRIEVAL SYSTEM
The information retrieval system of PDB files on NEC personal computer[9] was converted to these on the workstation. Using this function, a user can seek the file by author name, keyword, and amino acid sequence as retrieval tag, into all of the PDB's files. This system also has multi-windows and a graphical user interface similar to the display system.
The method of retrieval
In this system, retrieval keys are classified into three categories : "keywords" are the words included in HEADER, COMPND, and SOURCE records of PDB file, "author" is an author of a paper, "sequence" is an arrangement of an amino acid and nucleic acid. The process of retrieval is to compare the retrieval key with information included in the PDB files. To perform information retrieval efficiently, keyword, author, and sequence index files are made from the original data files of PDB. The extraction of information from PDB files is the same as the personal computer version[9] basically. Search by order is used as the method of retrieval. The retrieval is completed within two, one, or eight seconds in the case of search for a keyword, an author, or a sequence, respectively.
Outline of the retrieval operation
On the information retrieval by keyword or author, retrieval with right truncation matching as well as complete matching can be performed. These operations are done on the main retrieval panel as seen in Figure 12.
On the information retrieval by keyword or author, one can change input mode ( Keyword input mode / author input mode ) by toggle switch ( Figure 12-(a) ). If one inputs the target keyword or author name, the number of found files is displayed at the right side of the input line ( Figure 12-(b) ). When one adds wild card ( * ) to the input key, one can retrieve with the right truncation matching. Other operations are the same as the complete matching. If the matched keywords or names exist, each name and the number of files are listed on another panel. Then one can select target keyword ( s ) or name ( s ) for displaying the retrieved result or using the set operations.
On the information retrieval by sequence, the one-letter symbols of amino acids or bases is used as input sequence. Capital letters and small letters are distinguished, the former express the sequence of amino acids in a protein and the latter express the sequence of bases in a nucleic acid. If the target file is found, the number of files is displayed at the right side of the input line. In the case of information retrieval by keyword and author, the retrieved result is displayed on the window (Figure 13).
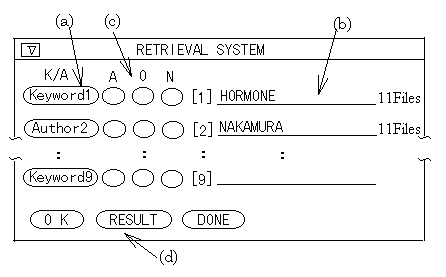
Figure 12 . Main retrieval panel. (a) The button to change input mode. (b) The input line. (c) The button to change the set operation mode. (d) The button to show the result panel.

Figure 13 . The retrieved result panel. (a) The number of found files. (b) The list of found files. (c) The HEADER, COMPND, and SOURCE data extracted from the original data file of PDB. (d) The overview button which can show record data (c) quickly. (e) The display buttons which can show the molecule on the display window or the contents of PDB file.
Set operation
The retrieved results are stored, therefore, the set operations are possible between them. On this operation, "AND", "OR", and "NOT " are possible between two retrieved results. One has only to push the appropriate button beside the input line : "A" (AND), "O" (OR), or "N" (NOT). ( Figure 12-(c) ).
AVAILABILITY
This program is available through the anonymous FTP site of The Chemical Software Society of Japan.
( http://cssj.chem.sci.hiroshima-u.ac.jp/ftp /indexe.html )
REFERENCES
1) Nakano, H., "Molecular Graphics", Science House Co., Ltd., Tokyo, (1987) (in Japanese).
2) Uno, T., Zhang, J., Sawano, T., Yamana, K. and Nakano, H., J. Chem. Software, 2, 212- 218 (1995) (in Japanese).
3) Nakano, H., Information, No.5, 91-99 (1986) (in Japanese).
Nakano, H., Information, No.10, 65-75 (1986) (in Japanese).
Nakano, H., Information, No.11, 77-84 (1986) (in Japanese).
Nakano, H., Information, No.12, 64-70 (1986) (in Japanese).
Nakano, H., Information, No.1,104-111 (1986) (in Japanese).
4) Ferrin, T. E., Huang, C. C., Jarvis, L. E. and Langridge, R., "The MIDAS display system", J. Mol. Graphics, 6, 13-27 (1988).
5) Kuznetsov, D. A., and Lim, H. A., "VisiCoor: A simple program for visualization of proteins", J. Mol. Graphics , 10, 25-28 (1992).
6) Evans, S. V., "SETOR: Hardware lighted three-dimensional solid model representations of macromolecules", J. Mol. Graphics, 11, 134-138 (1993).
7) Sayle, R. A., and Milner-White E. J., "RASMOL: Biomolecular graphics for all", Trends Biochem. Sci., 20, 374-376 (1995).
8) Bernstein, F. C., Koetzle, T. F., Williams, G. J. B., Meyer, E. F., Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T., and Tasumi, M., J. Mol. Biol. 112, 535-542 (1977).
9) Nakano, O., Nakano, H., Sasakura, T., Mukaida, K., Yamana, K., and Sangen, O., J. Association of Personal Computer for Chemists, 2, 141-150 (1991) (in Japanese).
Return