A Computational Chemical Investigation of the Dehydration
and Dehydrogenation of Ethanol on Oxide Catalysts
Yuji SHINOHARA, Tsuyoshi NAKAJIMA, Satoshi SUZUKI,
Shozi MISHIMA and Hideaki ISHIKAWA
 Return
Return
1. Introduction
The dehydration and dehydrogenation of alcohols on oxide catalysts have been studied as the fundamental catalytic reactions by many investigators. Generally, on acid catalysts, ethylene is formed by dehydration of ethanol (at a relatively low temperature, diethyl ether is formed), whereas acetaldehyde is formed by dehydrogenation on basic catalysts. Thus, these are used as test reactions to estimate the acidity and/or basicity of a solid catalyst [1].
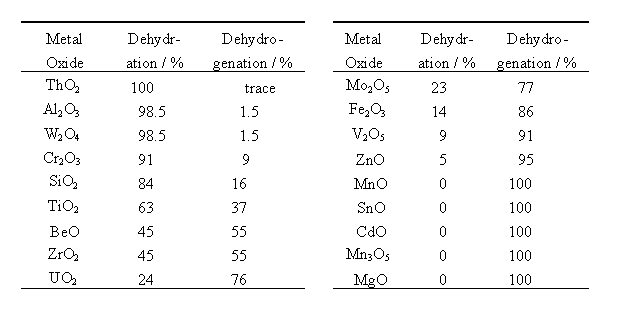
Sabatier and Mailhe [2] have found a series of oxide catalysts (shown in Table 1) in order of the selectivities of these reactions. Some parameters of the oxides, such as electronegativities of metal ions (hereafter denoted as M) [3], acidic and basic characteristics [4], parameters proposed by Eucken and Wicke [5], etc., were examined to explain the selectivity order of the catalysts and the reaction mechanisms. However, explanation using these parameters has merely achieved partial success and the molecular details of these reactions have not been fully understood.
Table 1 Selectivities of various metal oxides for catalytic dehydration and dehydrogenation of ethanol. (P. Sabatier and A. Mailhe)
For the dehydration of ethanol on Al2O3, a consecutive mechanism (Figure 1) including ethoxide formation has been proposed [6]. According to this mechanism, ethanol adsorbs on an acid site, i.e., a surface hydroxyl group, on the oxide surface and forms water and an ethoxide. A b hydrogen (hereafter denoted as Hb) of the ethoxide then shifts to the neighboring base site (oxide ion, hereafter denoted as O) and ethylene is formed.
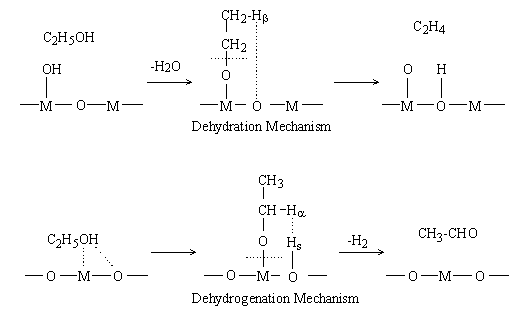
Fig. 1 "Mechanism I" of ethanol dehydration and dehydrogenation on oxides.
M : metal atom
For the dehydrogenation of ethanol, a mechanism (Figure 1) including ethoxide formation is also proposed [5]. According to the mechanism, ethoxide and a proton (hereafter denoted as Hs) are formed on the respective interaction of surface M and O with the hydrogen and oxygen atoms of the hydroxyl group of ethanol. Surface ethoxide and Hs thus formed are bound to M and O, respectively. An a hydrogen of ethoxide (hereafter denoted as Ha) shifts to Hs as a hydride ion, and acetaldehyde and molecular hydrogen are formed.
Eucken and Wicke proposed a concerted mechanism explaining the reaction selectivities of oxide catalysts by the sizes of M [5]. According to their mechanism (Figure2 (1)), when M is small, it is submerged under the oxide surface. The surface hydroxyl group (hereafter denoted as O-Hs) and O then interact concertedly with the oxygen (hereafter denoted as OE) and Hb of ethanol, respectively, and ethylene and water are formed. On the other hand, when M is large, it appears on the surface and interacts with Ha of ethanol, as shown in Figure 2 (2). Simultaneously, the O interacts with hydrogen (hereafter denoted as HE) of a hydroxyl group, and acetaldehyde is formed. In summary, there are two main mechanisms for the dehydration and the dehydrogenation of ethanol: Mechanism I (Figure 1) including ethoxide formation and Mechanism II (Figure 2) proposed by Eucken and Wicke. There is, however, no decisive evidence to show which mechanism is valid.
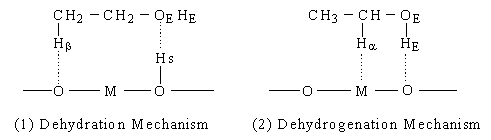
Fig. 2 "Mechanism II" of ethanol dehydration and dehydrogenation on oxides.
In the previous paper [7], we computed the interaction of ethanol with Bronsted or Lewis acid sites of a series of oxides using the DV-Xa method and examined the relationship between the computed results and the reaction selectivity. We found that the electron density of the ethanol oxygen in the HOMO corresponds well to the dehydration selectivity of the oxides in the case of ethanol interacting with the Bronsted site. In the system of ethanol and the Lewis site, we found that any population of the bonds of the system, including the population of the bond between the Lewis site and the ethanol oxygen, has no regular relationship with the dehydration selectivity of the oxides. However, the details after ethanol adsorption have remained unclear.
The purpose of this study was to examine the validity of Mechanisms I and II using the DV-Xa method. For this purpose, models of the transition states of the mechanisms were made and submitted to computation, and we then examined whether the two dependencies of the computed values of parameters and the reaction selectivities on the kind of oxides agreed well with each other. With regard to Mechanisms I and II, the electronic states of ethoxide interacting with surface O or Hs and of ethanol concertedly interacting with surface M and O were respectively computed and examined.
2. Calculation method
The DV-Xa cluster method, one of the density functional methods, runs much faster than any other ab initio method and is capable of computing large clusters containing heavy elements. Moreover, it is well-known that the method provides reliable values for many physical quantities except for the total energy of a system [8]. The computations were carried out using the general-purpose M-860 computer (made by the Hitachi Corporation) located in the General Information Processing Center of Shinshu University. For developing coordinate data and processing computed results, a personal computer PC-9801, made by the NEC Corporation, was used.
3. Transition state models of the reactions
3.1 The transition state model of Mechanism I
If the dehydration proceeds by Mechanism I, Hb of ethoxide shifts to O of oxides. Because the ethoxide is generally stable on oxides [6], it is reasonable to think that this shift is rate-determining. Transition state models of the shift were created for oxides of Si, Ti, Zn, Mn, Mg and Cd and are shown in Figure 3 (Models in the cases of SiO2 and MgO are shown as examples). To simplify the model, only one M was placed in the cluster of oxide, and seven atoms and ions (Hb, C, C, O, M, O, H) were placed in a plane. Experimental values from the literature [9] were adopted for bond distances and angles. The values for ethoxide were made equal to the experimental ones for ethanol [9]. Each terminal oxygen of the cluster was replaced by a hydroxyl group or water, and the system was kept electrically neutral. The coordination numbers of M and O were made equal to the actual numbers realized in the oxide crystals. Among the oxides of Ti, Mg, Mn, Cd, Si and Zn submitted to computations, the crystals of the former four oxides have the structure of rutile or NaCl type, where M's exist as six dentate centers. Therefore, we created a model including M coordinated by six ligands, i.e., hydroxyl groups and water, for each oxide. For the latter two oxides of Si and Zn whose crystals have tetrahedral structures, we created models including M's coordinated by four ligands. In other words, the system of ethoxide on SiO2 was modeled as Si(OH)3(OC2H5). Similarly, the other systems were modeled as Ti(OH)3(H2O)2(OC2H5), Zn(OH)(H2O)2(OC2H5), Mn(OH)(H2O)4(OC2H5), Mg(OH)(H2O)4(OC2H5) and Cd(OH)(H2O)4(OC2H5). On the basis of the results of structural optimizing of the models by MOPAC, bond angles in C2H5O-M-OH (M=Ti, Mg, Mn and Cd) were made equal to 109.45 as is the case for the models of ethoxides on oxides of Si and Zn. The structure parameters adopted for the models are listed in Tables 2 and 3.
According to Mechanism I, the dehydrogenation proceeds when Ha of an ethoxide shifts to Hs of a surface hydroxyl group at the transition state. The shift of Ha is thought to be rate-determining as is the case for dehydration. Therefore, the transition state of the reaction was delineated by the model shown in Figure 4 (the cases of SiO2 and MgO as examples). Six atoms and ions (Ha, C, O, M, O, Hs) were placed in a plane, and computations were carried out for models of six oxides as is the case for dehydration.
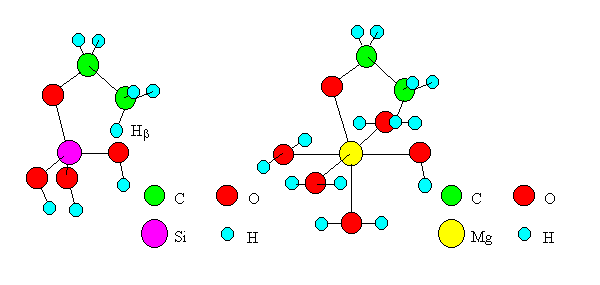
Fig. 3 Transition state models of dehydration by "Mechanism I".
Table 2 Geometrical parameters (M-O bond distance).
| Metal | Si | Ti | Zn | Mg | Mn | Cd
|
| d (M-O) /A | 1.61 | 1.95 | 1.97 | 2.11 | 2.21 | 2.35
|
Table 3 Geometrical parameters.

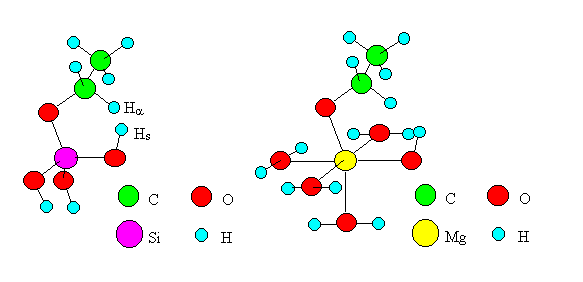
Fig. 4 Transition state models of dehydrogenation by "Mechanism I".
3.2 The transition state model of Mechanism II
For the dehydration on oxides by Mechanism II (Figure 2) [5] of Eucken and Wicke, transition state models including one M per model were created (Figure 5, the cases of SiO2 and MgO are shown as examples). Bond angles of O-M-O (one of the two O's interacts with Hb of ethanol) were made equal to 90 (oxides of Ti, Mg, Mn and Cd) or 109.5 (oxides of Si and Zn). The other bond angles were made equal to those in the models of Mechanism I. The bond distance of the O-Hb bond was made equal to 0.957A, which is that of the O-H bond of water, and eight atoms and ions (M, O, Hs, OE, C, C, Hb, O) were placed in a plane. The computations were carried out for the models of six oxides as in the case of Mechanism I.
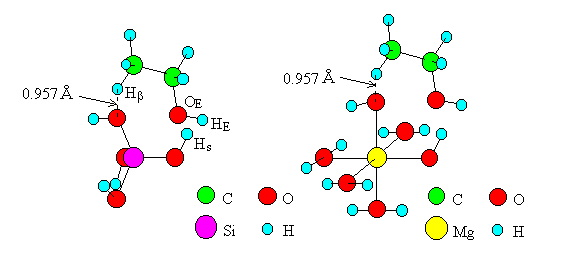
Fig. 5 Transition state models of dehydration by "Mechanism II".
For the dehydrogenation on oxides by Mechanism II, transition state models including one M were created (Figure 6, the cases of SiO2 and MgO are shown as examples), where six atoms and ions (M, O, HE, OE, C, Ha) were placed in a plane after the model of the dehydration. The surface of the oxide SiO2 was modeled as Si(OH)3, and those of the other oxides were modeled in the same way as Ti(OH)3(H2O)2, Zn(OH)(H2O)2, Mn(OH)(H2O)4, Mg(OH)(H2O)4, and Cd(OH)(H2O)4. The bond length of the HE-O bond was made equal to 0.957A, which is the distance of the O-H bond of water.

Fig. 6 The transition state model of dehydrogenation by "Mechanism II".
4. Calculation results and discussion
4.1 Mechanism I
4.1.1 Transition state model of the dehydration
Populations of the bonds in ethoxide in the transition state model of the dehydration are shown in Figure 7. The metal oxides are arranged on the abscissa in the order in which the selectivity decreases from left to right. The selectivity value is shown in the figure using the right ordinate. A number on the left ordinate indicates the population of a bond of ethoxide by setting that of the corresponding bond of free ethanol as a standard. For the Hb-O bond which has no correspondence to the free ethanol, the population was plotted as computed. The population, derived from Mulliken's population analysis, of 1s orbitals of atoms A and B is defined as 2S(CA1CB1 + CA2CB2), where CAi and CBi are the coefficients of the molecular orbital for the i-th electron and S is the overlap integral. Therefore, the value is a certain measure of the strength of the bond. Thus, when a value on the left ordinate is positive, the bond of the ethoxide is stronger than the corresponding bond of free ethanol. Inversely, when negative, the bond is thought to be weaker.

When ethanol is dehydrated to ethylene via the transition state shown in Figure 3, it is favorable that the C-Hb bond in ethoxide become weaker, whereas the Hb-O bond becomes stronger under the action of O of the oxide. As seen from Figure 7, the higher the dehydration selectivity of the oxide is, the larger is the population of the Hb-O bond, whereas the smaller that of the C-Hb bond is. Therefore, the transition state shown in Figure 3 is reasonable on the basis of the experimentally determined selectivity order of the oxides. There is no relation between the selectivity order and dependency of the populations of the other bonds on oxides. These facts indicate that there is no tendency of shifts in the electronic state of ethoxide itself to affect the dehydration selectivity. It is likely that shifts in the electronic state of ethoxide to facilitate the dehydration appear when Hb interacts with a base site on the oxides.
4.1.2 The transition state model of the dehydrogenation by Mechanism I
The transition state model of the dehydrogenation shown in Figure 4 was submitted to computation and the results are shown in Figure 8. The number on the left ordinate indicates the population of a bond of ethoxide by setting that of the corresponding bond of free ethanol as a standard. For the M-O and Ha-Hs bonds, which have no correspondence in the free ethanol, populations were plotted as computed.

For the dehydrogenation to proceeds according to Mechanism I, it is favorable that the C-Ha bond of the ethoxide is weak ,whereas the Ha-Hs bond is strong. Therefore, if Mechanism I is valid, it is expected that the population of the Ca-Ha bond is smaller, whereas the population of the Ha-Hs bond is larger on the oxide with higher dehydrogenation selectivity. As seen from Figure 8, however, the population of the C-Ha bond did not have the expected tendency. This fact indicates that the dehydrogenation selectivity is not affected by the kind of oxides. Sedlacek has computed the model (PrO)M(OH) (M=Al, Sc, Cr; Pr=2-propyl) of the dehydrogenation of 2-propanol on oxides by the CNDO/2 method [10]. He has compared the electric charge on an atom and the bond order in the 2-propyl group with those of the corresponding atom and bond in free 2-propanol. He found that Ha in the 2-propyl group is more negative than that in the free molecule and that the bond order of the C-Ha bond in the 2-propyl group is smaller than that in the free molecule. The bond order was scarcely dependent on the kind of oxide. From these findings, he has concluded that there is no parameter capable of explaining the dehydrogenation selectivities of the oxides. In the present study, the oxide-dependent shifts of populations of bonds in ethoxide at the transition state of the dehydrogenation are small and are about 15 % of the shifts in the case of the dehydration. This fact indicates that the ease of dehydrogenation is independent of the kind of oxide. Therefore, selectivity of the dehydration or dehydrogenation is thought to be controlled by the ease of the dehydration. In other words, the reason the dehydrogenation is dominant on some oxide is that the dehydration is difficult on the oxide.
4.2 Mechanism II
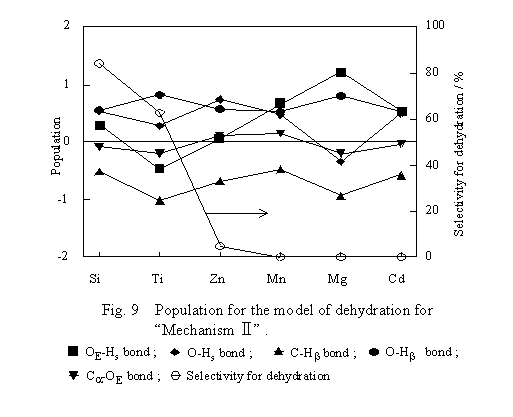
4.2.1 The transition state model of the dehydration
The transition state model of Mechanism II shown in Figure 5 was submitted to computation of bond populations, and the results are shown in Figure 9. As for the populations of the C-Hb and C-OE bonds, differences in those between the transition state and free ethanol were plotted. The populations of the other bonds were plotted as computed. For the dehydration to proceed in accordance with Mechanism II, it is favorable that the C-Hb bond of ethanol becomes weak, whereas the OE-Hs bond becomes strong on interaction of ethanol with the surface. Except for the case of SiO2, the OE-Hs bond is weaker on the oxides with higher dehydration selectivity. The O-Hb bond is strong and the C-Hb bond is weak irrespective of the kind of oxide. The result indicates the ease of the shift of Hb to O on all oxides and is inconsistent with the dependency of the dehydration selectivity on the kind of oxide. Moreover, the O-Hs bond is strong irrespective of the kind of oxide except for MgO, and population shift of the C-OE bond is small compared with the others. These facts imply that the ethanol dehydration proceeds in accordance with some mechanism other than Mechanism II.
4.2.2 The transition state model of the dehydrogenation
The transition state model of Mechanism II shown in Figure 6 was submitted to computation of bond populations, and the results are shown in Figure 10. As for the populations of the C-Ha, C-OE and OE-Ha bonds, differences in those between the transition state and free ethanol were plotted. The other populations were plotted as computed. For the dehydrogenation to proceed in accordance with Mechanism II, it is favorable that the C-Ha and OE-HE bonds of ethanol are weak, whereas the Ha-M and O-HE bonds are strong at the transition state.

As seen from Figure 10, however, there is no bond formation between Ha and M. Both of the C-OE and OE-HE bonds are strong irrespective of the kind of oxide, and the population shifts of these bonds are inconsistent with the dehydrogenation selectivities of the oxides. These facts indicate that HE and Ha are not captured by the oxides. Thus, there is no favorable change for dehydrogenation in the strength of the bonds of ethanol at the transition state of Mechanism II and, therefore, it is concluded that the ethanol dehydrogenation proceeds in accordance with some mechanisms other than Mechanism II.
To summarize, it is the transition state model of Mechanism I including the ethoxide formation that successfully explains the order of dehydration selectivity of the oxide, not the concerted Mechanism II proposed by Eucken and Wicke. Therefore, it is concluded that the ethanol dehydration proceeds according to Mechanism I and that the selectivity is determined by the ease of the Hb shift to an oxide ion on the oxides.
5. Conclusions
The electronic states of ethoxide and ethanol interacting with oxide surfaces were computed by the DV-Xa method and the following conclusions were obtained.
1. In the case of the ethanol dehydration, the computed results for the transition state model of Mechanism I successfully explain the order of the dehydration selectivities of the oxides, whereas the computed results based on Mechanism II scarcely explain the order. It is concluded that the ethanol dehydration and dehydrogenation proceed according to the consecutive mechanism including ethoxide formation.
2. On the oxide with higher dehydration selectivity, the bond order between Hb of the ethoxide and the surface oxide ion is higher, whereas that of the C-Hb bond of ethoxide is lower. These results imply that the dehydration selectivity of an oxide catalyst is controlled by the ease for Hb to shift to the oxide ion on the catalyst.
3. The population shifts of the bonds of ethoxide are small in absolute values and the tendency of the shifts is inconsistent with the dependency of the dehydrogenation selectivities on the oxides. The reason why the ethanol dehydrogenation proceeds on basic oxides is that the dehydration is suppressed rather than that the dehydrogenation is accelerated on the oxides.
6. Reference
1) T. Imanaka, Shokubai, 25, 474 (1983).
2) P. Sabatier et A. Mailhe, Ann. de Chem. et de Phys., 20, 289 (1910).
3) Shokubai Gakkai, "Gensobetu Shokubai Benran", Chijin Shokan, Tokyo (1978), p255.
4) K. Tanaka and K. Tamura, Bull. Chem. Soc. Jpn., 37, 1862 (1964).
5) ref.3), p790.
6) H. Arai and Y. Saito, Y. Yoneda, Bull. Chem. Soc. Jpn., 40, 312 (1967); H. Arai, J. Take, Y.
Saito and Y. Yoneda, J. Catal., 9, 146 (1967); ibid., 10, 128 (1968); Y. Murakami,
T. Hattori, Kougyoukagaku, 70, 2098 (1967); T. Hattori and K. Shirai, Y. Murakami,
Shokubai, 22, 40 (1980).
7) Y. Shinohara, H.Satozono, T. Nakajima, S. Suzuki and S. Mishima, J. Chem. Software, 4, 41 (1997).
8) H. Adachi,"Ryoushi Zairyou Kagaku Nyuumon", Sankyo Shuppan, Tokyo (1991);
M. .Morinaga, N. Yukawa and H. Adachi, J .Phys .Soc. Jpn., 53, 653 (1984); J. Kawai, K. Maeda, M. Takami, Y. Muramatsu, T. Hayashi, M. Motoyama and Y. Saito, J. Chem. Phys., 98, 3650 (1993); H. Adachi, S. Shiokawa, M. Tsukada, C. Satoko and S. Sugano, J. Phys. Soc. Jpn., 47, 1528 (1979); M. Tsukada, C. Satoko and H. Adachi, J. Phys. Soc .Jpn.,47, 1610 (1979); M. Tsukada, C. Satoko and H. Adachi, J. Phys. Soc. Jpn.,48, 200 (1980).
9) Nippon Kagaku Kai, "Kagaku Benran�$Bqk�(B�9�", Maruzen, Tokyo (1966), p1203; K. Tanabe, T.
Aoyama and K. Fueki, "Kinzoku Sankabutu to Hukugou Sankabutu", Kodansha, Tokyo (1978), p42; Samsonof, "Saishin Sankabutsu Benran", Nisso Tsuushinsha, Tokyo (1970), p15.
10) J. Sedlacek, J. Catal., 44, 318 (1976).
Return