A Study on Binding Modes of Nonsteroidal Anti-inflammatory Drugs to COX1 and COX2 as Obtained by Dock4.0
Eiichi AKAHO, Chikako FUJIKAWA, Howell I. RUNION, Craig R. HILL and Hidehiko NAKANO
 Return
Return
This paper was presented at 1999 Dock user group meeting, "DOCK: Applied Structure-Based Drug Design" held on April 16-17, at University of California at San Francisco, USA.
1 Introduction
With an advance of computer technology and protein science, automated docking programs have been developed and utilized in various fields[1 - 9]. One of the most old and well-known docking programs is Dock4.0[10]. The dock program in general is designed to find favorite orientation of a ligand in a receptor. A typical receptor might be an enzyme with a well-defined active site. The ligand structure may be taken from the crystal structure of the ligand-enzyme complex or can be drawn manually by using a chemical modeling software. The orientation of the ligand is evaluated with a shape scoring function and/or a function approximating ligand-enzyme binding energy. The shape scoring function is an empirical function resembling the van der Waals attractive energy. The ligand-enzyme binding energy is taken to be approximately the sum of van der Waals attractive, van der Waals repulsive, and coulombic electrostatic energy as shown below[10]:
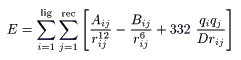
where each term is a double sum over ligand atoms i and receptor atoms j, Aij and Bij are van der Waals repulsion and attraction parameters, rij is the distance between atoms i and j, qi and qj are the point charges on atoms j and j, D is the dielectric function, and 332 is a factor that converts the electrostatic energy into kilocalories per mole.
It is said that Dock4.0 among other docking programs is a so-called standard program in pharmaceutical companies for general lead compound development[11]. However, due to its complexity in nature, difficulties have been encountered. Upon the release of Dock4.0 in May 1997, the authors obtained it and installed it on SGI(O2) : Model;R5000SC(180MHz).
Cyclooxygenase, also known as prostaglandin endoperoxide syntheses (pHs) or COX, is the key enzyme of the biosynthetic pathway leading to the formation of prostaglandins. It has recently been reported that this enzyme exists in two isoforms[12]. The amino acid compositions of the two isoforms are about 60% identical and their affinity towards arachidonic acid, the natural substrate, appears to be quite similar[13].
Prostaglandins responsible for inflammatory process can be sufficiently controlled with nonsteroidal anti-inflammatory drugs (NSAIDs). On the other hand, NSAIDs produce adverse effects on the gastrointestinal (GI) mucosa, kidney, and homeostasis limiting their clinical utility. The role of COX1 is thought to be a production of "housekeeping" prostaglandin critical to autocrine / paracrine responses to circulating hormones, and maintenance of normal renal function, gastric mucosal integrity, and homeostasis[14]. COX2 is thought to contribute to the increase in prostaglandins observed in inflammed tissues.
Therefore, anti-inflammatory drugs, which selectively inhibit COX2 without affecting an enzyme activity of COX1, would be an ideal anti-inflammatory drug. Binding modes of COX1 and COX2 to anti-inflammatory drugs must be different, but there are no reports to show the binding modes of existing nonsteroidal anti-inflammatory drugs against COX1 and COX2. Thus an attempt was made to examine those binding modes of NSAIDs against COX1 and COX2 by utilizing Dock4.0.
2 Background
in vitro studies of COX1 and COX2 using murine enzymes have been reported [15, 16]. Meade et al. reported differential activity of seven NSAIDs on COX1 and COX2 which were expressed in and isolated from Cos 1 cells. They measured IC50 which represented the NSAIDs concentration required to inhibit enzyme activity by 50%. 6-MNA (the active metabolite of nabumetone) exhibited the lowest ratio of IC50 (COX2 / COX1) which was 0.1. This indicates that nabumetone is roughly 7 times more active on COX2 than on COX1. They found that sulindac and indomethacin were the least selective to COX2.
Barnett et al studied differential activity of various NSAIDs on human COX2 / COX1 expressed in and purified from baculovirus [17]. They found that the IC50 ratios (COX2 / COX1) of indomethacin, anirolac, flurbiprofen, ketoprofen, suprofen, naproxen, diclofenac, ibuprofen, fenclofenac, nabumetone(6-MNA), meclofenamate, nimesulide, NS-398, and niflumic acid were 14.7, 12.8, 12.7, 4.6, 4.0, 3.3, 1.6, 0.6, 0.5, 0.3, 0.03, 0.02, 0.02, and 0.008 respectively. This study shows that indomethacin, anirolac and flurbiprofen were fairly COX1 specific drugs. On the other hand, nabumetone (6-MNA), meclofenamate, nimesulide, NS-398, and niflumic acid were more specific inhibitors for COX2. Based on these reported in vitro studies on differential activity of various NSAIDs, grouping of NSAIDs in terms of COX1 / COX2 selectivity was performed. Indomethacin, sulindac and flurbiprofen were classified as COX1 selective NSAIDs. Nabumetone (6-MNA), meclofenamate, and niflumic acid were classified as COX2 selective NSAIDs. The remaining NSAIDs, naproxen, diclofenac, and ibuprofen were termed as equipotent NSAIDs.
3 Methodology
The known structures of NAISDs were drawn by InsightII (Molecular Simulation Incorporated) and the NSAIDs were used as ligands for docking study. COX1 and COX2 structures were obtained from Brookhaven protein databank (URL: http://www.pdb.bnl.gov/pdb-bin/pdbmain). After removing water molecules and assigning hydrogens and charges, computer docking was performed. The results were evaluated according to the proposed rule mentioned in the Background section.
4 Results and Discussion
Evaluation of docking results will be performed based on steric complementarity. This can be called the best fitting of ligand to the enzyme pocket. There is no universal rule to judge which one exhibits the best steric and concrete complementarity. At this point it is pointed out that the following three factors are primarily involved to influence binding conformation between a ligand and a protein. These are binding energy, hydrogen bonding, and hydrophobic-hydrophobic interaction. The binding energies of hydrogen bonds range from 1 - 7 kcal/mol while that of hydrophobic-hydrophobic interactions among methylene groups is estimated to be 0.37 kcal/mol[18]. Therefore, the former is, on the average, about 10 times stronger than the latter. It is possible to identify the existence of hydrogen bonding quantitatively in the docked conformation by using the chemical software InsightII, but this program does not quantify that of hydrophobic-hydrophobic interactions well. Taking these factors into consideration we applied the following criteria to judge the relative binding selectivity of ligands to COX1 and COX2. We do not wish to establish that these are the absolute rules by which to judge the best binding mode of a particular ligand to a particular protein but that these criteria can be used to compare the binding selectivity of ligands to similar but different types of proteins as presented in the current study.
- the one whose number of H-bonds is greatest (e.g.2) with the lowest binding energy
- the one whose number of H-bonds is greatest (e.g.2) with the next lowest binding energy (When necessary, Step 2 is repeated in the order of decreasing binding energy to the one with the same number of H-bonds.)
- the one whose number of H-bonds is next greatest (e.g.1) with the lowest binding energy
- the one whose number of H-bonds is next greatest (e.g.1) with the next lowest binding energy(When necessary, Step 4 is repeated in the order of decreasing number of H-bonds and binding energy.)
Based on the above parameters an evaluation of the docked results of each ligand against COX1 and COX2 was performed. Location as well as the distance of hydrogen bonding were identified in terms of the relevant atoms of the amino acid residue and the ligand.
4. 1 Docking mode of COX2 selective NSAIDs against COX
Based on the proposed criterion stated in the Results and Discussion section COX2 selective NSAIDs were evaluated as shown in Table 1.
Table 1 Docking results of COX2 selective NSAIDs
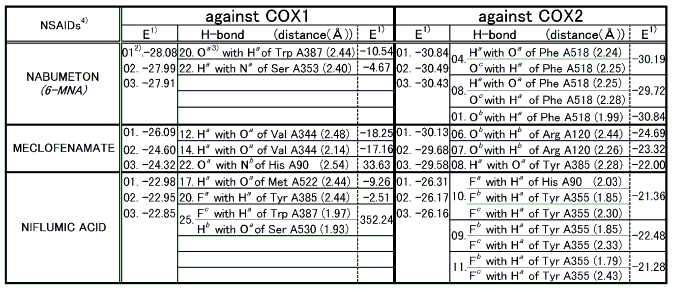
Note: 1) E represents total binding energy between a ligand and a protein and is given in kcal/mol.
2) Dock results are numbered (01) to (25) with the lowest energy being the first(01).
3) Details for superscripts, a, b,and c are explained in Figure 1.
4) NSAID represents Nonsteroidal Antiinflammatory Drug.
- Upon the computer docking of nabumetone against COX2 nabumetone, as in the docking result No.04, exhibited two H-bonds, one between the carboxyl hydrogen of nabumetone and the carbonyl oxygen of phenylalanine (residue A518) (distance: 2.24A), and the other between the carboxyl oxygen of nabumetone and the amine hydrogen of phenylalanine (residue A518) (distance: 2.25A)(Figure 2). The docked binding energy was -39.19 kcal/mol. On the other hand, upon the computer docking of nabumetone against COX2 as shown in the docking result No.20 nabumetone exhibited just one H-bond between the methoxy oxygen of nabumetone and the indole NH hydrogen of tryptophane (residue A387) (distance:2.44A)(Figure 3). The docked binding energy was -10.54 kcal/mol. This indicates that nabumetone inhibited COX2 with more affinity and strongly than COX1: in other words, nabumetone can be said to be COX2 selective.
- Upon the computer docking of meclofenamate against COX2, meclofenamate as in the docking result No.06 exhibited one H-bond between the carbonyl oxygen of meclofenamate and the amide hydrogen of arginine (residue A120) (distance: 2.44A)(Figure 4). The docked binding energy was -24.69kcal/mol. On the other hand, upon the docking of meclofenamate against COX1, meclofenamate as in the docking result No.12 exhibited one H-bond between the carboxyl hydrogen and the carbonyl oxygen of valine (residue A344) (distance: 2.48A)(Figure 5). The docked binding energy was -18.25 kcal/mol. This indicates that meclofenamate inhibited COX2 more firmly and strongly than COX1: in other words, meclofenamate can be said to be COX2 selective.
- Upon the computer docking of niflumic acid against COX2, niflumic acid as in the docking result No.10 exhibited three H-bonds; one between the fluorine atom of niflumic acid and the imidazole 1-NH hydrogen of histidine (residue A90) (distance: 2.03A), and another between the fluorine atom of niflumic acid and the hydroxyl hydrogen of tyrosine (residue A355) (distance: 1.85A), and the other between the fluorine atom of niflumic acid and the hydroxyl hydrogen of tyrosine (residue A355) (distance: 2.30A)(Figure 6). The docked binding energy was -21.36 kcal/mol. On the other hand, upon the computer docking against COX1, niflumic acid as in the docking result No.17 exhibited one H-bond between the amine hydrogen of niflumic acid and the carbonyl oxygen of methionine (residue A522) (distance: 2.44A)(Figure 7). The docked binding energy was -9.26 kcal/mol. This indicates that niflumic acid inhibited COX2 with more affinity and strongly than COX1: in other words, niflumic acid will be said to be COX2 selective.
An overview of COX2 binding mode to COX2 selective NSAIDs showed that there existed one to three H-bonds with the net total being at least six when NSAIDs such as nabumetone, meclofenamate, niflumic acid, indomethacin, sulindac, and flurbiprofen were bound to COX2. Amino acid residues involved in such hydrogen bonds were Phe A518, Arg A120, Tyr A385, His A90, and Tyr A355. Phe A518 and His A90 were reported by R. G. Kurumbail et al. [12] but the rest of the amino acid residues were not.
4. 2 Docking mode of COX1 selective NSAIDs against COX
Based on the proposed criterion COX1 selective NSAIDs were evaluated as shown in Table 2. Upon the computer docking evaluation of COX1 selective NSAIDs, one showed a good correlation with the experimental result while another did not and a third mediocre conformity[13].
Table 2 Docking results of COX1 selective NSAIDs
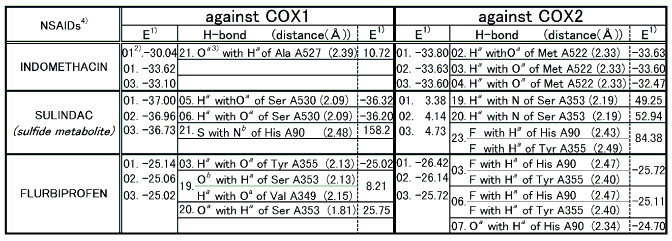
Note: 1) E represents total binding energy of a protein and is given in kcal/mol.
2) Docking results are numbered (01) to (25) with the lowest being the first(01).
3) Details for superscripts, a, b,and c are explained in Figure 8.
4) NSAID is Nonsteroidal Antiinflammatory Drug.
- The best possible docking mode of indomethacin against COX1 was identified to be the docking result No.21 in which one H-bond was observed between the carbonyl oxygen of indomethacin and the amine hydrogen of alanine (residue A527) (Figure 9). The H-bond distance was found to be 2.39A. The docked binding energy was 10.72 kcal/mol. On the other hand, the best possible docking mode of indomethacin against COX2 was identified to be the docking result No.02 (Figure 10). One H-bond existed between the carboxyl hydrogen of indomethacin and the carbonyl oxygen of methionine (residue A522) with its distance being 2.33A. The docked binding energy was -33.63 kcal/mol.
- The best possible docking mode of sulindac against COX1 was identified to be the docking result No.05 in which one H-bond was observed between the carboxyl hydrogen of sulindac and the hydroxyl oxygen of serine (residue A527) (Figure 11). The H-bond distance was found to be 2.09A. The docked binding energy was -36.32 kcal/mol. On the other hand, the best possible docking mode of sulindac against COX2 was identified to be the docking result No.19 (Figure 12). One H-bond existed between the carboxyl hydrogen of sulindac and the amine nitrogen of serine (residue A353) with its distance to be 2.19A. The docked binding energy was 49.25kcal/mol.
- The best possible docking mode of flurbiprofen against COX1 was identified to be the docking result No.03 in which one H-bond was observed between the carboxyl hydrogen of flurbiprofen and the hydroxyl oxygen of tyrosine (residue A355) (Figure 13). The H-bond distance was found to be 2.13A. The docked binding energy was -25.02 kcal/mol. On the other hand, the best possible docking mode of flurbiprofen against COX2 was identified as shown in the docking result No.03 (Figure 14). Two H-bonds existed; one between the fluorine atom of flurbiprofen and the imidazole 1-NH hydrogen of histidine (residue A90) with its distance to be 2.47A, and the other between the fluorine atom of flurbiprofen and the hydroxyl hydrogen of tyrosine (residue A355) with its distance being 2.40A. The docked binding energy was -25.72 kcal/mol.
4. 3 Docking mode of Equipotent NSAIDs against COX
The docked results of the remaining NSAIDs are shown in Table 3. Naproxen showed a COX2 selectivity. Although the docking modes against either COX1 or COX2 exhibited two H-bonds, the total binding energy for a COX2 docking was lower than that for COX1 showing its COX2 selectivity. Diclofenac also exhibited a COX2 selectivity. The number of H-bonds with COX2 was two, while that with COX1 was one. The total docked binding energy was lower in the COX2 docking than in the COX1 docking confirming its COX2 selectivity. Ibuprofen, on the other hand, showed COX1 selectivity. No H-bonds was observed in the COX2 docking while one H-bond in the COX1 docking. Since the one with H-bond is ranked higher than the one without H-bond, ibuprofen was identified to be COX1 selective. Therefore, no docking consistency was observed in this group. This is what was expected, because this group is mediocre and it showed that these have neither COX1 nor COX2 selectivity.
Table 3 Docking results of equipotent NSAIDs
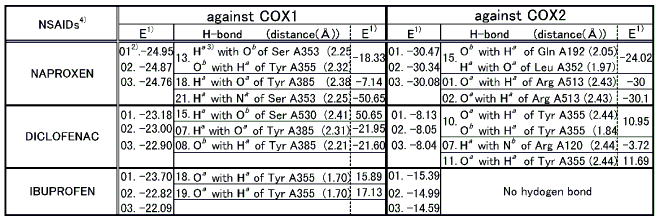
Note: 1) E represents total binding energy between a ligand and a protein and is given in kcal/mol.
2) Dock results are numbered (01) to (25) with the lowest energy being the first(01).
3) Details for superscripts, a, b,and c are explained in Figure 15.
4) NSAID represents Nonsteroidal Antiinflammatory Drug.
4. 4 Overall docking mode of NSAIDs against COX
An overview of COX2 binding mode to COX2 selective NSAIDs indicated that one to three H-bonds were formed between the inhibitor and the enzyme, with the net total being at least six. Amino acid residues involved in such hydrogen bonds were Phe A518, Arg A120, Tyr A385, His A90, and Tyr A355. Further examination of these hydrogen bonds showed that a net total of at least twelve hydrogen bonds existed when NSAIDs such as nabumetone, meclofenamate, niflumic acid, indomethacin, sulindac, and flurbiprofen were bound to COX2. Amino acid residues involved in such hydrogen bonds were Phe A518, Arg A120, Tyr A385, His A90, Tyr A355. Met A522, Ser A353, Gln A192, Leu A352, and Arg A513. . Phe A518 and His A90 were reported previously(12) but the rest of the amino acid residues have not been. The binding mode of COX2 selective NSAIDs resembled the result reported by R.S. Spangler [13], that is, three out of three COX2 selective NSAIDs showed the same selectivity reported experimentally [13]. An overview of COX1 binding mode to COX1 selective NSAIDs indicated that one to three hydrogen bonds were formed with the net total being at least four. Amino acid residues involved in such hydrogen bonds were Ala A527, Try A355, Ser A353, and Val A349. As far as the binding mode of COX1 selective NSAIDs against COX1 is concerned, one conformed to the in vitro study reported by R.S. Spangler, another did not conform at all and a third presented mediocre conformity.
5 Conclusions
Using Dock4.0 on SGI O2 workstation, it was shown that the binding mode of COX2 selective NSAIDs coincided with the results reported in in vitro study by R.S. Spangler [13]. Thus, it can be said that there was a fairly good correlation between the Dock4.0 results and the reported in vitro study. As far as the binding mode to COX1 selective NSAIDs is concerned one corresponded to the in vitro study reported by R.S. Spangler another did not at all and a third presented a fair fit. Although some correlation was shown to exist between the docked results and the reported experimental results, further studies will be needed to substantiate the existence of correlation between the docked and experimental results. It was shown that there existed one to three hydrogen bonds with the net total being at least twelve when inhibitors such as nabmetone, meclofenamate, niflumic acid, indomethacin, sulindac, and flurbiprofen were bound to COX2. Amino acid residues involved in such hydrogen bonds were Phe A518, Arg A120, Tyr A385, His A90, Tyr A355. Met A522, Ser A353, Gln A192, Leu A352, and Arg A513. Phe A518 and His A90 were reported by R. G. Kurumbail et al [12] but the rest of the amino acid residues have not been reported previously. This may indicate that the binding of these agents is more complex that previously thought.
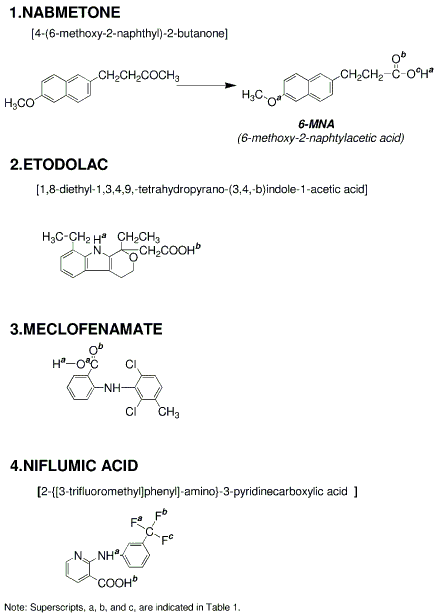
Figure 1. COX2 selective NSAIDs and atoms responsible for H-bonding
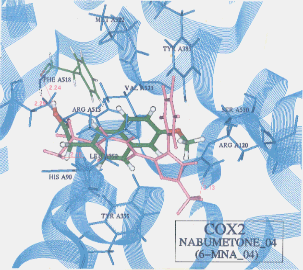
Figure 2. Complex between nabumetone and COX2 as obtained by Dock4.0
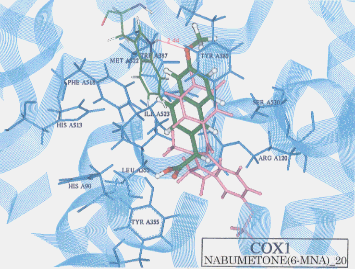
Figure 3. Complex between nabumetone and COX1 as obtained by Dock4.0
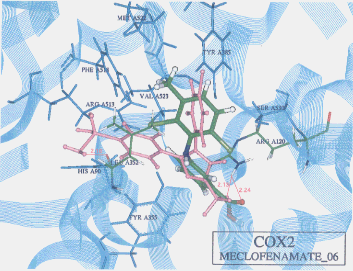
Figure 4. Complex between neclofenamate and COX2 as obtained by Dock4.0
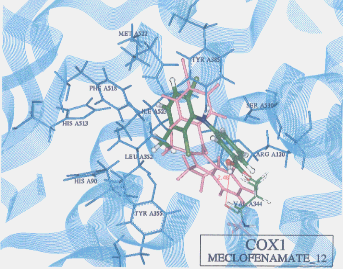
Figure 5. Complex between neclofenamate and COX1 as obtained by Dock4.0
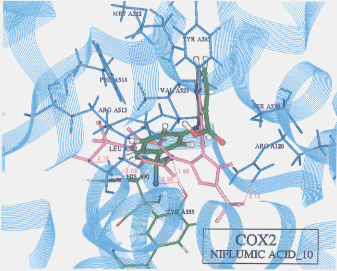
Figure 6. Complex between niflumic acid and COX2 as obtained by Dock4.0
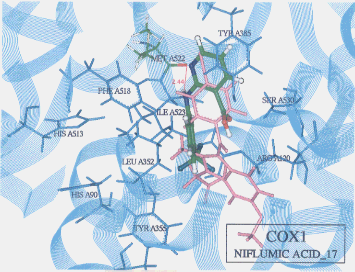
Figure 7. Complex between niflumic acid and COX1 as obtained by Dock4.0
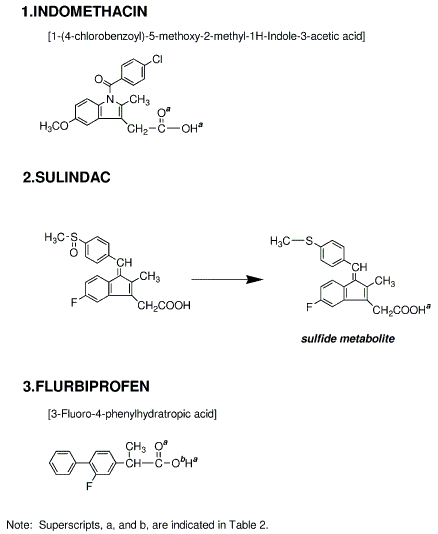
Figure 8. COX1 selective NSAIDs and atoms responsible for H-bonding with COX
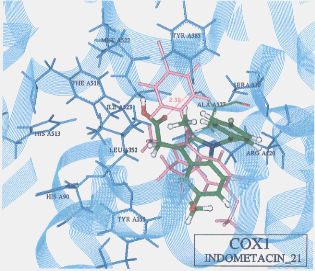
Figure 9. Complex between indomethacin and COX1 as obtained by Dock4.0
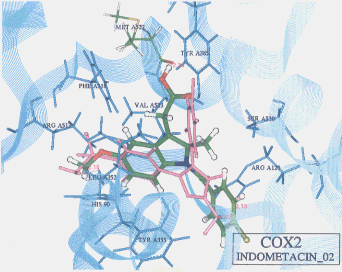
Figure 10. Complex between ndomethacin and COX2 as obtained by Dock4.0
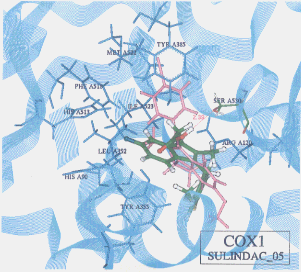
Figure 11. Complex between sulindac and COX1 as obtained by Dock4.0
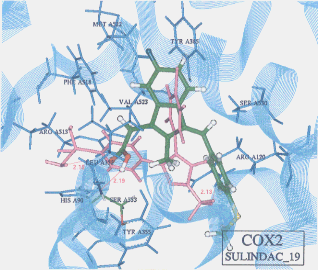
Figure 12. Complex between sulindac and COX2 as obtained by Dock4.0
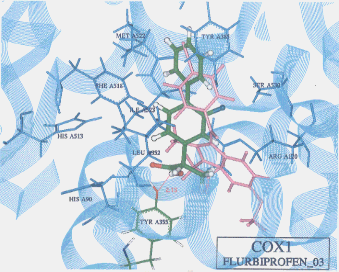
Figure 13. Complex between flurbiprofen and COX1 as obtained by Dock4.0
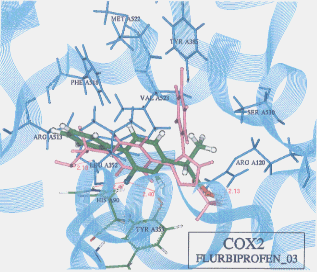
Figure 14. Complex between flurbiprofen and COX2 as obtained by Dock4.0
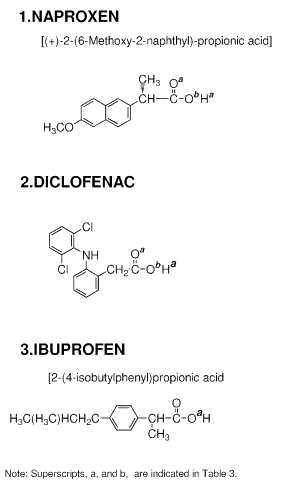
Figure 15. Equipotent NSAIDs and atoms responsible for H-bonding with COX
This work was supported in part by a 1999-2000 International Cooperation Study, Scientific Grant of the Ministry of Education and the authors express their sincere appreciation to the Ministry of Education. This work was also supported in part by a 1999-2001 Interdisciplinary Study Grant A of Kobe Gakuin University and the authors are grateful to the president of Kobe Gakuin University.
References
[ 1] DesJarlais, R. L., Seibel, G. L., Kuntz, I. D., Furth, P. S., Alvarez, J. C., Ortiz de Momtellano, P. R., DeCamp, D. L., Babe, L. M. and Craik, C. S., Structure-based design of nonpeptide inhibitors specific for the human immunodeficiency virus 1 protease, Proc. Natl. Acad. Sci., 87, 6644-6648 (1990).
[ 2] Meng, E. C., Shoichet, B. K. and Kuntz, I. D., Automated Docking with Grid-Based Energy Evaluation, Journal of Computational Chemistry, 13, 505-524 (1992).
[ 3] Lewis, R. A., Roe, D. C., Huang, C., Ferrin, T. E., Langridge, R. and Kuntz, I. D., Automated site-directed drug design using molecular lattices, J. Mol. Graphics, 10 (1992).
[ 4] Ring, C. S., Sun, E., McKerrow, J. H., Lee, G. K., Rosenthal, P. J., Kuntz, I. D. and Cohen, F. E., Structure-based inhibitor design by using protein models for the development of antiparasitic agents, Proc.Natl.Acad.Sci., 90, 3583-3587 (1993).
[ 5] Meng, E. C., Gschwend, D. A., Blaney, J. M. and Kuntz, I. D., Orientational Sampling and Rigid-Body Minimization in Molecular Docking, PROTEINS: Structure, Function, and Genetics, 17, 266-278 (1993).
[ 6] Milne, G. W. A., Nicklaus, M. C., Driscoll, J. S. and Wang, S., National Cancer Institute Drug Information System 3D Database, J. Chem. Inf. Comput. Sci., 34, 1219-1224 (1994).
[ 7] Meng, E. C., Kuntz, I. D., Abraham, D. J. and Kellogg, G. E., Evaluating docked complexes with the HINT exponential function and empirical atomic hydrophobicities, Journal of Computer-Aided Molecular Design, 8, 299-306 (1994).
[ 8] Grootenhuis, P. D. J., Roe, D. C., Kollman, P. A. and Kuntz, I. D., Finding potential DNA-binding compounds by using molecular shape, Journal of Computer-Aided Molecular Design, 8, 731-750 (1994).
[ 9] Roe, D. C. and Kuntz, I. D., BUILDER v.2: Improving the chemistry of a de novo design strategy, Journal of Computer-Aided Molecular Design, 9, 269-282 (1995).
[ 10] Kuntz, I. D., "Dock4.0," University of California at San Francisco, 1998
[ 11] Personal communication with a computer drug design scientist in one of the Japanese leading pharmaceutical Companies (Dec.1998)
[ 12] Kurumbail, R.G., Stevens, A. M., Gierse, J. K., McDonald, J. J., Stegeman, R. A., Pak, J. Y., Gildehaus, D., Miyashiro, J. M., Penning, T. D., Seibert, K., Isakson, P. C., and Stallings, W. C., Structural basis for selective inhibition of cyclooxygenase-2 by anti inflammatory agents, Nature, 384, 644-648 (1996).
[ 13] Pedretti, A., Villa, A. M., Villa, L. and Giulio, V., INTERACTIONS OF SOME PGHS-2 SELECTIVE INHIBITORS WITH THE PGHS-1: AN AUTOMATED DOCKING STUDY BY BIODOCK, IL FARMACO, 52(6-7), 487-491 (1997).
[ 14] Spangler, R. S., Cyclooxygenase 1 and 2 in Rheumatic Disease: Implications for Nonsteroidal Anti-inflammatory Drug Therapy, Seminars in Arthritis and Rheumatism, 26, 435-446 (1996).
[ 15] Loll, P.J., Piccot, D., Ekabo, O., Garavito, R.M., Synthesis and use of iodinated nonsteroidal anti-inflammatory drug analogs as crystallographic probes of the prostaglandin H2 synthase cyclooxygenase active site, Biochemistry, 35, 7330 (1996).
[ 16] Meade, E.A., Smith, W.L., DeWitt, D.L., Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs, J Biol Chem., 286, 6610-6614 (1993).
[ 17] Barnett, J., Chow, J., Ives, D., et al., Purification, characterization and selective inhibition of human prostaglandin G/H synthase 1 and 2 expressed in the baculovirus system, Biochim Biophys Acta, 1209, 130-139 (1994).
[ 18] Remington's Pharmaceutical Sciences, Ed. by Gennard AR, Mack Publishing Co., Pennsylvania (1985).
Return