 Return
Return
The recent development of the Internet, especially World Wide Web (WWW), has changed the computer network and computer graphics dramatically. For three-dimensional (3D) graphics on the computer, the Virtual Reality Modeling Language (VRML) is used on the WWW. Molecular graphics using VRML has also been developed recently [5,6]. This new technique enables us not only to display molecules on the screen but also to change the point of view with respect to the object shown.
Under such circumstances, we have
developed an interactive molecular modeling and molecular graphics
program MOLDA for Windows, which is designed to be used as an
interface to the ab initio MO program GAUSSIAN 94 [7] and the
MD simulation program AMBER 4.0 [8] as well as the molecular mechanics
program MM2 [4]. The 3D molecular structures in VRML format can
be generated by using this program and the molecular models are
exported over the Internet by using VRML.
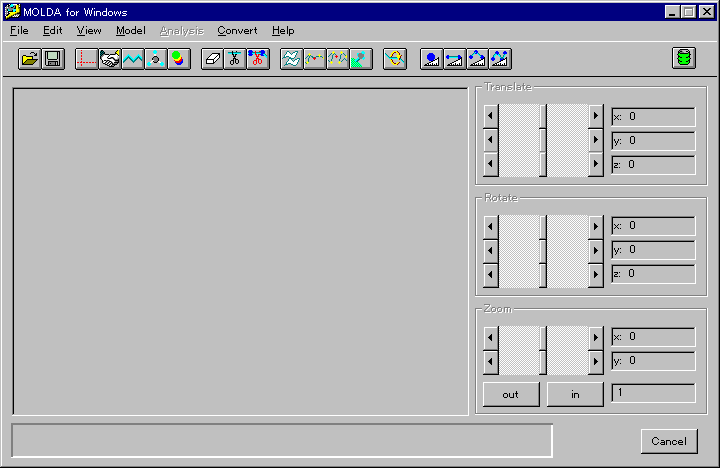
When MOLDA is running, all of the
commands of MOLDA (summarized in Table 1) are allocated to the
menu bar (Fig. 1). The main feature of the molecular modeling
is the same as that of MOLDA4 described previously [3] except
for the use of a mouse pointing device. Using the molecular modeling
functions of MOLDA, the labour involved in preparing molecular
structure data for molecular science is considerably reduced.
Selection of a menu activates the corresponding subcommands. Menus
are listed below.
1. File Menu
The 'File'
menu deals with molecular coordinate file input and output. The
valid molecule file format is MODRAST/MOLDA[3,9] and MSC's
XMol XYZ file format [10]. If no file format is specified, MODRAST/MOLDA
file format is assumed by default. Molecular coordinates can
be saved in VRML file format (See section 3.2.).
2. Edit Menu
The 'Edit'
menu deals with the pasting of the Cartesian coordinates obtained
from GAUSSIAN 94 or MM2. 'Undo' and 'Copy' commands are also available.
3. View Menu
The 'Edit'
menu deals with the change of the screen appearance (such as showing
x- and y-axes, scaling molecular size and locating the number
on each atom).
Table 1. Menus/Submenus and commands in MOLDA for Windows
| Menu | Submenu | Option | Description | |
|---|---|---|---|---|
| File | ||||
| Open | Read molecular structure data in MOLDA or XMol file format | |||
| Save | Save molecular structure data in MOLDA or XMol file format | |||
| Save As | Save molecular structure data in MOLDA or XMol file format as specified file name | |||
| Save in VRML | Save molecular structure data in VRML file format ( Dreiding sticks, ball-and-stick and spacefilling models are available) | |||
| Hardcopy of display | ||||
| Exit | Exit MOLDA for Windows | |||
| Edit | ||||
| Undo | Undo delete | |||
| Copy | Copy MOLDA data to clipboard | |||
| Paste | ||||
| MOLDA | Paste MOLDA data in clipboard on MOLDA | |||
| MM2 | Paste MM2 data in clipboard on MOLDA | |||
| GAUSSIAN | Paste GAUSSIAN data in clipboard on MOLDA | |||
| View | ||||
| Axes | Show x- and y-axes | |||
| Atom Number | Locate the number on each atom | |||
| Auto Scaling | Scale the size of molecule automatically | |||
| Cancel Atoms Automatically | Cancel duplicated atoms automatically when merged | |||
| Cancel Bonds Automatically | Cancel duplicated bonds automatically when merged | |||
| Model | ||||
| Input | ||||
| Atom | Input the number of atoms, the Cartesian coordinate and the atomic numbers | |||
| Bond | Generate a bond between specified two atoms | |||
| Alkane | Make the all-trans conformation of an n-alkane molecule by input of the number of carbon atoms | |||
| Hydrogen | Add hydrogen atoms to the carbon atoms | |||
| a-No | Replace a specified atom by another element | |||
| Cancel | ||||
| Atom | Delete a specified atom | |||
| Bond | Delete a specified bond | |||
| Component | Delete a specified group of atoms | |||
| Merge | ||||
| Point | Place a molecule by specification of an atom so that the specified atom is at the specified position | |||
| Atom | Replace an atom or group of atoms of the molecule by another molecule | |||
| Bond | Connect two molecules in such a way that their specified bonds overlap | |||
| Subst | Substitute an atom or a group of atoms with a common substituent | |||
| Group | ||||
| Cn | Do operation Cn | |||
| m | Do operation s | |||
| i | Do operation i | |||
| Sn | Do operation Sn | |||
| Internal Rotation | Do an internal rotation around a specified bond | |||
| Move | ||||
| Atom | Move the origin to a specified atom | |||
| Bond | Move the origin to the midpoint of two specified atoms | |||
| Analysis | ||||
| Coordinate | Give coordinates of a specified atom | |||
| Distance | Calculate distance between two specified atoms | |||
| Angle | Calculate bond angle | |||
| Dihedral Angle | Calculate dihedral angle | |||
| Convert | ||||
| MM2 -> MOL | Convert MM2 output into MOLDA data | |||
| AMBER -> MOL (Movie) | Convert AMBER trajectory file into MOLDA data as movie | |||
| MOL -> MM2 | Convert MOLDA data into MM2 input data | |||
| MOL -> GAUSSIAN | Convert MOLDA data into GAUSSIAN 94 input data | |||
| Help | ||||
| Info | Show information of this program |
Fig. 1 A menu screen of MOLDA for Windows.
4. Model Menu
The 'Model' menu deals with the construction of molecular models. The input of molecular structure is commenced with the 'Input' submenu. Input data are the Cartesian coordinates of atoms, the atomic numbers and atom connection lists, which are easily generated with mouse operation. The all-trans conformation of an n-alkane molecule can be generated in a one-step operation by using the 'Alkane' command. The input structure need only be a carbon skeleton, as hydrogen atoms can be added to the skeleton by using the 'Hydrogen' command. A specified atom may be replaced by another element by using the 'a-No.' command.
The 'Merge' submenu enables any molecules made by MOLDA to be connected with each other in several modes (a specified spatial arrangement, a substitution, a ring fusion, a bridged ring-connection and a spiro ring-connection) by mouse operation.
Another interesting feature of MOLDA
is the 'Group'
submenu. By using the commands in the 'group'
submenu, the corresponding 'symmetry
operation' can be performed.
The symmetry operations are defined as follows: operation 'C6z',
for example, is defined so that the molecule has a C6
symmetry axis around the z-axis. Thus, the amount of input data
may be reduced. It is worth mentioning that the construction of
a complicated molecule such as coronene is greatly simplified
by using this operation (Fig. 2).
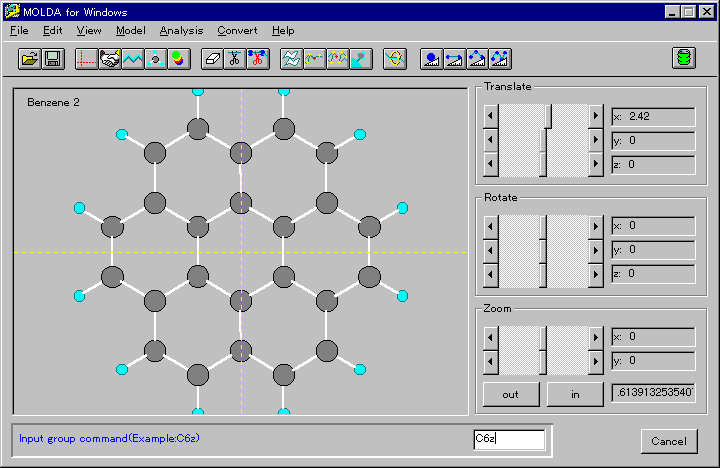
Fig. 2 A coronene molecule generated by using C6 operation.
5. Analysis Menu
The 'Analysis'
menu deals with the calculation of bond distances, bond angles
and dihedral angles of a molecule.
6. Convert Menu
The 'Convert'
menu deals with the conversion of the Cartesian coordinates obtained
by using several molecular science programs into MOLDA format
file. The details are described in section 3.3.
3.2. The Feature of Molecular Graphics
The 3D molecular structures in VRML format can be generated by using MOLDA. These molecular structure data created by MOLDA or converted from GAUSSIAN 94, AMBER 4.0 and MM2 can be viewed in 3D by using a VRML viewer and demonstrated on WWW. At present, Dreiding-sticks (Fig. 3), spacefilling spheres (Fig. 4) and ball-and-stick models (Fig. 5) are available. The molecular models written in VRML can be easily operated by the VRML viewer not only by local users but also by remote users, who can use terminal computers connected to the Internet. Moreover, the molecular models can be viewed platform-independently; in other words, they can be displayed on the DOS/V, Macintosh and Unix machines.
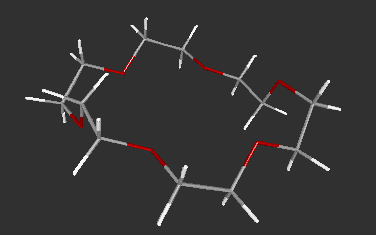
Fig. 3 A Dreiding-stick model of 18-crown-6.
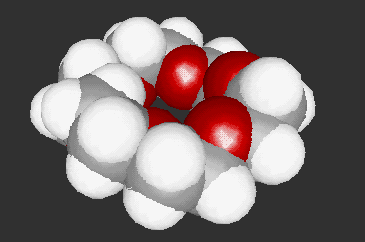
Fig. 4 A space-filling model of 18-crown-6.
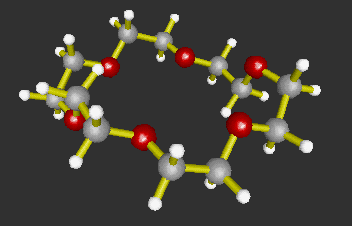
Fig. 5 A ball-and-stick model of 18-crown-6.
3.3. The Feature of an Interface to Molecular Science Programs
When the 'Convert'
menu is selected, the data format for MOLDA and several molecular
science programs is converted to each other as described below.
1. MOLDA can make input data for
molecular mechanics (MM2) or ab initio MO calculations (GAUSSIAN
94). For MM2, the conversion of data for MOLDA into those for
MM2 was described in detail elsewhere [3].
2. The output files of GAUSSIAN 94,
AMBER 4.0 and MM2 can be converted to the data of MOLDA format.
The results of MD simulations can be animated on the screen.
Return