 Return
Return
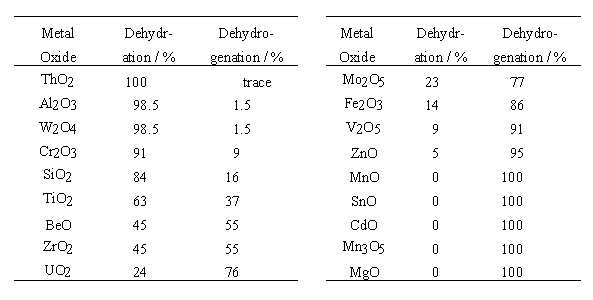
In the table, the dehydration selectivity is defined as the percentage of ethanol changed into water and ethylene, and the dehydrogenation selectivity as the percentage of ethanol changed into hydrogen and acetaldehyde.
Let us assume that there exists a series of catalysts arranged in a certain order of experimentally determined selectivity for a given reaction. The quantum chemical computations are then carried out on a postulated transition state of the reaction. If the postulated one is valid, computed physical quantities, such as strength of a bond being broken or formed at the transition state, should change according to the catalyst order. On the other hand, if the postulated transition state is invalid, change in the calculated physical quantity should be independent of the catalyst order. Therefore, in the case of the reaction for which a series of catalysts is experimentally known to have an ordered catalytic property, it is considered possible to inspect the validity of the reaction mechanism by quantum chemically computing the transition state expected from the mechanism.
In this study, models consisting of B or L sites on oxides of several metals in the 3rd - 5th periodic groups and ethanol were computed for the first time using an ab initio method. From these results, it was investigated whether the dehydration and the dehydrogenation selectivities were determined by the electronic state of ethanol interacting with acid sites. It was also determined whether a population, a measure of strength of a covalent bond, changes in agreement with the catalyst series of the dehydration selectivity. From these inspections, the validity of the proposed mechanisms for the dehydration and the dehydrogenation was discussed.
The DV-Xa method employed here is one of the ab initio methods and it is possible to significantly reduce its cpu time compared to the other ab initio methods. It is also possible to deal with much larger clusters involving heavy atoms. It is well known that the method supplies reliable values for many physical quantities except for the total energy of the system[11].
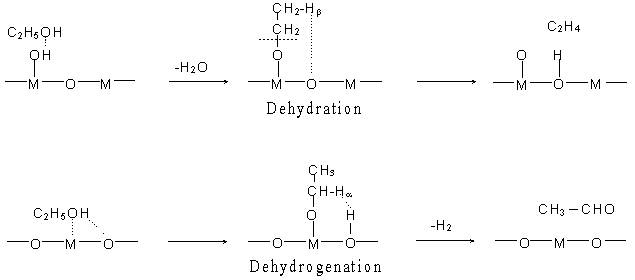
Fig. 1 Reaction mechanisms of ethanol dehydration and dehydrogenation on metal oxides.
M = Metal ion
In this study, interactions of ethanol with the B or L sites were studied. The B sites are the protons of surface hydroxyl groups and the L sites are the surface metal ions [8, 15]. The interactions are the first step of the reactions where an alcohol interacts with the acid sites and produces either a carbenium ion or an ethoxyl group.
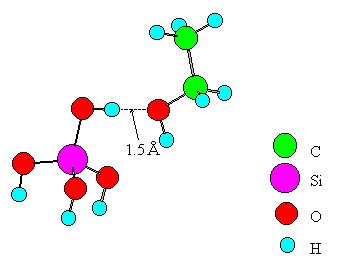
Fig. 2 A model of ethanol interacting with a Broensted acid.
Only one metal ion was placed in the cluster of an oxide for simplicity. Experimental values from the literature [16, 17, 18, 19] were adopted for bond distances of ethanol and the oxide. The distance between the B site and the oxygen of ethanol is set to 1.5A for the following reasons. The stable structure of the B site (SiO2-Al2O3, SiO2, ZnO, MgO, CdO) - ethanol system was calculated using the MOPAC-PM3 method in the range of 2.5~1.0A for the distance. For the case of SiO2-Al2O3 and SiO2, which exhibits the highest dehydration selectivity, the system had the highest stability at the distance of 1.8A. On the other hand, systems involving the other oxides had no stable structures. The systems were therefore compared to each other at the distance of 1.5A where the interaction must be stronger than that at 1.8A. Each terminal oxygen of the cluster was replaced by a hydroxyl group or water, and the system electrically was kept neutral. The coordination number of a metal ion or an oxide ion was set to the practical numbers realized in the oxide crystals. The central metal ion in the cluster was Si, Ti, Zn, Mg, Mn, or Cd. A similar model was made for the case of SiO2-Al2O3 and subjected to the computation. In the model, SiO2 was approximated by Si(OH)4. Similarly, TiO2, ZnO, MgO, MnO2, CdO, and SiO2-Al2O3 were approximated by Ti(OH)4(H2O)2, Zn(OH)2(H2O)2, Mg(OH)2(H2O)4, Mn(OH)2(H2O)4, Cd(OH)2(H2O)4, and Si(OH)3-OH-Al(OH)3, respectively.
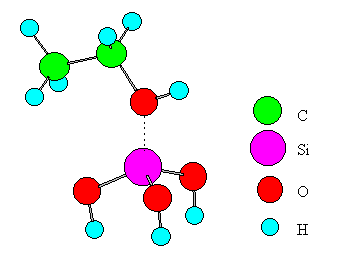
Fig. 3 A model for ethanol interacting with a Lewis acid.
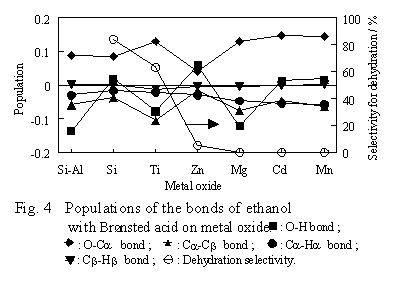
The metal oxides were arranged on the abscissa in order of the dehydration selectivity experimentally determined by P. Sabatier and A. Mailhe, i.e., in the order where the selectivity decreases from left to right. The selectivity value is shown in the figure by using the right ordinate. The numbers on the left ordinate indicate the population of a bond of ethanol interacting with the B site, by setting that of the corresponding bond of free ethanol as a standard. The population used here is derived from Mulliken's population analysis and shows overlap of the orbitals of the two bonding atoms. Therefore, the value is a certain measure of the strength of the (covalent) bond. That is, when a value on the left ordinate is positive, the bond of the ethanol interacting with the B site is stronger than the corresponding bond of free ethanol. Inversely, when negative, the bond is thought to be weaker. When ethanol interacts with the B site and is dehydrated to ethylene, it is favorable that the O-H and Ca -Cb bonds in ethanol become stronger and that the Ca -O bond becomes weaker. As shown in the figure, the population of each bond significantly varies from one oxide to another. However, the variation has no relation to the order of the dehydration selectivity of the oxides. For example, the population of the Cb-H bond, which breaks in the case of ethylene formation, has a similar value for every oxide, which exhibits very different dehydration selectivity. These results indicate that the ethylene formation by fission of the Cb-H bond is not facilitated even if ethanol interacts with the B site.
When ethanol interacts with the B site, the population of the O-Ca bond is positive for any oxide. This result means that the bond becomes stronger with the interaction. On the other hand, the population of the Cb-H bond is negative, so the bond becomes weaker during the interaction. It may be impossible, however, to discuss the reaction mechanisms from these results for the two bonds, because absolute values of the populations and their variations with the kind of metal oxides are too small.
Figure 5 shows the computed populations of the bond between ethanol oxygen and the B site formed during the early stage of the dehydration.
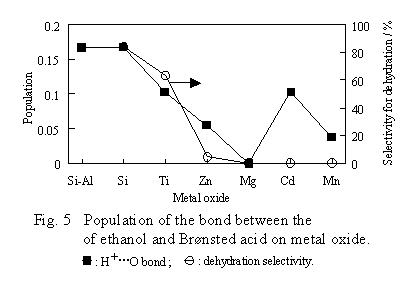
Except for the case of CdO and MnO, there is a close relationship between the population and the dehydration selectivity. This relationship means that the dehydration becomes favorable when the bond between the B site and the ethanol oxygen becomes stronger. This result agrees with the E1 mechanism, i.e., the proton moves from the B site to the ethanol oxygen in the first stage of the dehydration, and then the protonated species is dehydrated to an ethyl cation, which converts into ethoxide by reacting with surface oxygen. The generation of the ethoxide is rate-determining. For the case of CdO and MnO, there is no correlation between the population and the dehydration selectivity. This is likely caused by using 1.5A which could be unsuitable for the two oxides as the distance between the B site and the ethanol oxygen, and that oxide surfaces could not be sufficiently represented by such small clusters as our computation models.
In the field of theoretical organic reactions, the electronic density in the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) is widely used as an important index of reactivity. Therefore, the electronic density and orbital composition of HOMO of the transition state of the dehydration were examined. As a result, it was found that the main component of HOMO was the orbitals of the oxygens of ethanol and the surface hydroxyl group. In Figure 6, the electronic density of the ethanol oxygen in HOMO was plotted against the kind of oxides.
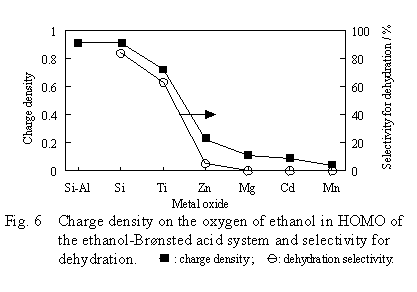
The obtained curve corresponded well to the variation in dehydration selectivity. This result means that the higher the electronic density of the ethanol oxygen in HOMO becomes, the higher the dehydration selectivity.
The population of each bond of ethanol which interacts with the Lewis acid site is shown in Figure 7.
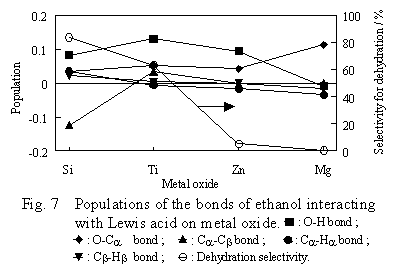
In this system, the absolute value of the population of the bond building ethanol was bigger than that computed for the B site - ethanol system, because the metal ion interacts directly with the ethanol oxygen. Furthermore, the population varied greatly with the kind of oxides. The variations in the populations of the O-H and the Ca-Cb bonds were especially large. However, the trend in the variation was independent of the dehydration selectivity of the oxides. On the other hand, the strength of the O-Ca bond had a trend to become stronger by the interacting of ethanol with the acid site on the oxides. However, this trend is likely within the error range of the computation, as is the trend in the case of ethanol interacting with the B site.
The population of the bond between the metal ion and the ethanol oxygen is plotted in Figure 8 against the kind of oxides, together with the dehydration selectivity.
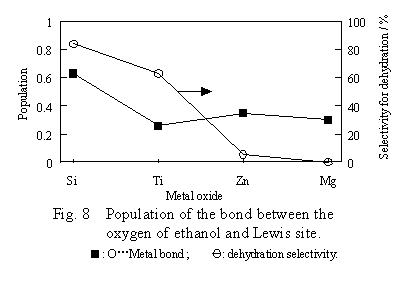
In contrast to the case of the B site, the population has no correlation to the dehydration selectivity of the oxide.For example, although the dehydration selectivity of titanium dioxide is high, the bond of the titanium ion with the ethanol oxygen is weak. Incidentally, in contrast to the case of the B site, the main component of the orbital near the HOMO of the L site system was not the orbital of the ethanol oxygen and of the surface hydroxyl group. Therefore, the electronic density of ethanol oxygen in HOMO could not be computed.
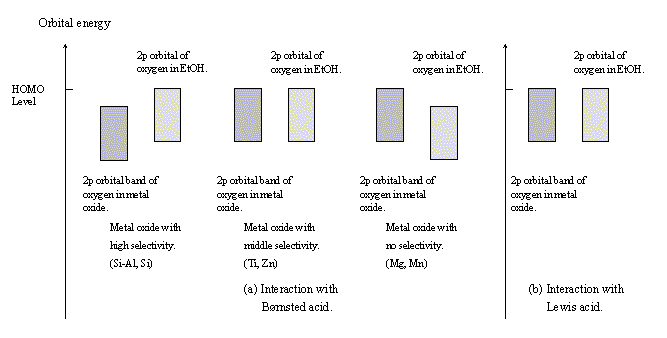
Fig. 9 Energy band structures of the calculated clusters.
(MgO, MnO, etc.) with no dehydration selectivity. The energy level of the 2p orbital of the ethanol oxygen was lower than that of the oxygen 2p band (HOMO of the system) of the metal oxide. With regard to CdO, although the dehydration selectivity was low, the band structure was similar to that of the oxides with the highest selectivity. This is likely caused by the fact that the computation model used was too small and too simple. Anyway, it was impossible to determine the right mechanism based on the discussion of the band structures, because the interaction of ethanol with the B site was thought to take place via the following two routes: the first is the route where a proton moves from an oxygen of the oxide to the ethanol oxygen, and the second is the route where some electrophile attacks the ethanol oxygen and then the dehydration occurs.
The band structure of the L site - ethanol system is schematically shown in Figure 9(b). The kind of oxide had no substantial influence on the band structure. The 2p orbital of the ethanol oxygen made an energy band, which agreed with that of the 2p band of the oxygen of the oxides. From these results, it was suggested that the electronic state of the oxygen of ethanol interacting with the L site was more similar to that of oxygen of the oxide than to that of the hydroxyl oxygen of ethanol.
It was also suggested that the electronic state of the ethanol oxygen was essentially the same for any oxide. Therefore, from the point of view of computational chemistry, the adsorption state of ethanol is nearly identical on any oxide. The dehydration selectivity of each oxide is, however, quite different from that of another oxide. Therefore, it is reasonable to conclude that the dehydration of ethanol does not occur on the L site.
Return