A Study on Docking Mode of HIV Protease and Their Inhibitors
Eiichi AKAHO, Garret MORRIS, David GOODSELL, David WONG and Arthur OLSON
 Return
Return
1 Introduction
Structural information of biological macromolecules complexed with ligands is being used to the extended degree in the discovery of novel pharmaceutical compounds. This information is crucial, and during the drug design cycle, experimental structures for a target protein complexed to ligand(s) can afford researchers to examine the detailed picture of the binding site and to design more active analogues [1]. A number of computer docking softwares have been utilized successfully to fulfill the purpose of this computer-based drug design [2]. This type of softwares is useful in evaluating the binding mode of candidate ligands or drug molecules to target macromolecules such as DNA, RNA, or enzyme. During the drug design cycle, experimental structures for a target protein complexed to ligand can allow scholars to examine the details of the binding site and to design de novo compounds [2].
There are two types of computer docking systems. The first is the direct type in which a user moves a ligand to a known active site and lets a docking program find a position of the one with the least Energy. In those positions hydrogen bonding and/or steric complimentarity usually take place [3, 4]. The second is an unbiased type in which a user does not have to direct a ligand to an active site, and can place a ligand in a random manner. The system finds an optimal position in which a ligand and a macromolecule bind.
AutoDock3.0 is an example of unbiased type and its version 3.0 has a ligand mobilized by a generic algorithm method and evaluates a rapid grid-based energy. Previous applications of AutoDock3.0 include the prediction of substrate binding to enzymes [5], computer-aided drug design of non-peptide inhibitors of HIV protease [6], and docking of two proteins [7]. Considerable efforts have been made to investigate an active inhibitor against HIV protease [8 - 10]. To date none of them are powerful enough to be successfully utilized therapeutically. Candidate inhibitors have been synthesized and the Ki was measured for each of 12 different inhibitors. Computer dockings between inhibitors and HIV protease by using AutoDock3.0 were performed on UNIX machines. Upon analyzing docking results of candidate inhibitors, the other preferred candidate compounds were proposed. It is said that the level of involvement in the elucidation of structures of HIV protease and its complexes with various inhibitors is enormous and numerous inhibitors have been reported [9].
The human immunodeficiency virus protease (HIV protease) has been a target for anti-HIV protease drug development in recent years. In other words, the world-wide AIDS epidemic has stimulated a research effort directed toward identifying therapeutic strategy effective for control of the disease and its causative agent. One approach of intense scrutiny over the past few years has been inhibition of the virally encoded protease, an enzyme essential for mutation of viral particles to their infectious stage [11] Attempts have been made to investigate various inhibitors such as nonpeptide sulfonamides, and fullerene [12, 13].
There have been varied attempts in the HIV protease related inhibitor studies such as that of the monocyte to chemokinase through CD4 signal dependent chemokinase receptor down-regulation by HIV protease -1 envelope gp20 [14], of chemoattractant receptor expression by HIV protease -1 envelope gp41 [15], and of HIV protease -1 expression by orphan upload receptor oligonucleotides [16]. This indicates that a variety of approaches should be taken into consideration in the inhibition of HIV protease infection.
2 Methodology
Complex structure of HIV protease with inhibitor 1 (inh1) was taken from the Internet (URL* http://www.rcsb.org/pdb/index.html). Various derivatives (first inhibitor group) of ih1 were synthesized and their Ki values against HIV protease were determined in vitro. For the docking of other inhibitors, the mode of interaction of inh1 against HIV protease was used as a standard docked model, the one used for RMSD calculation.
AutoDock3.0 was run by feeding an inhibitor and HIV protease. Since AutoDock3.0 consists of three major components, AutoGrid, AutoTors, and AutoDock3.0, the grid map was created by running AutoGrid as the first step. AutoTors was then run by assigning the root position and the number of torsions. Finally AutoDock3.0 was run in order to investigate mode of interaction between the inhibitor and HIV protease. Innovative inhibitors were proposed based on the docked result of the first inhibitor group.
3 Background
In order to compare the Gibbs free energy calculated from the experimental value with the AutoDock3.0 result, the following steps were applied to obtain the total Gibbs AutoDock3.0 free energy:
1) The torsional entropy was obtained by multiplying the number of torsions by the degree of freedom. This scoring function is based on the principles of QSAR (quantitative structure-activity relationships) and was parameterized using a large number of protein-inhibitor complexes for which both their structure and inhibition constants, or Ki, were known [17].
torsional entropy = number of torsions × degree of freedom ............(1)
2) The total Gibbs free energy was then obtained by taking into consideration the three factors, torsional entropy, lowest AutoDock3.0 energy obtained, and internal energy of inhibitor
Total AutoDock3.0 Gibbs free energy = torsional entropy +
lowest AutoDock3.0 energy - internal energy of inhibitor ...............(2)
On the other hand, experimental Gibbs free energy(DG) was obtained as follows:
DG = -RT × Ln Ki .............................................(3)
where,
R = (8.314/4.184 × 1000) J/Kmol
T = 298.2 °K
Ki in Mol
4 Results and Discussion
Figure 1 shows inhibitors synthesized [18] and tested for docking against HIV protease. Inh1 is a standard docking model ligand and when it is compared with the docking of other inhibitors one can tell how closer to the reference inhibitor the other inhibitors are docked. Upon the examination of docking feature between inh1 and HIV protease it was found that two types of hydrogen bonds were formed. As shown in Figure 2, one of the hydrogen bonds was found between gem-idol and an aspartate (residue number B25) moiety of HIV protease and the other between amide oxygen attached to proline ring and the other aspartate (residue number A25) moiety of HIV protease. Since 13 torsion angles were detected in inh1 the torsional entropy was obtained to be 4.05 Kcal/mol by multiplying it by the degree of freedom that was 0.3113 Kcal/mol (Table 1). Since the lowest total interaction energy of the complex and the internal energy for inh1 were -11.74 Kcal/mol and 4.03 Kcal/mol respectively, DGAutoDock was determined to be -11.99 Kcal/mol taking into account the torsional entropy by the use of equation (2).
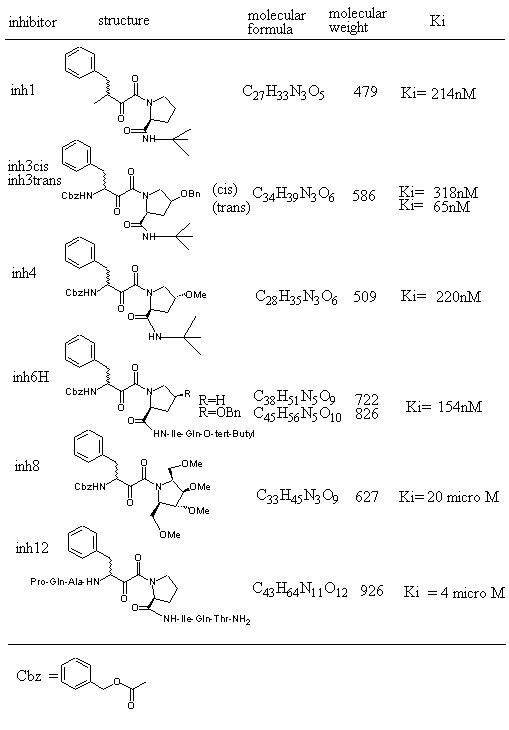
Figure 1. HIV protease inhibitors and their properties tested for AutoDock3.0 runs.
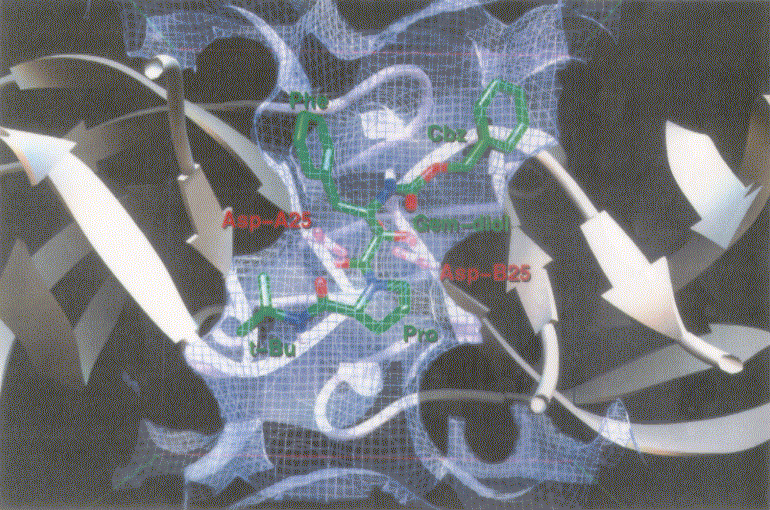
Figure 2. Docking mode of inhibitor 1 (inh1) as a reference inhibitor against HIV protease as obtained by AutoDock3.0. Atoms in inh1 are shown by different colors (red, oxygen; blue, nitrogen; and green, carbon).
Table 1. Comparison of experimental and computed Gibbs free energies of tested HIV protease inhibitors.
| inhibitor | number of torsions | torsional entropy (a) | total interaction energy of complex (b) | internal energy of inhibitor (c) | DGAutoDock (a+b-c) | DGexperimental |
|---|
| inh1 | 13 | 4.05 | -11.74 | 4.03 | -11.99 | -9.12 |
| inh3cis | 16 | 4.98 | -12.1 | 5.11 | -12.13 | -8.86 |
| inh3trans | 16 | 4.98 | -10.27 | 5.95 | -11.24 | -9.8 |
| inh4 | 14 | 4.36 | -11.19 | 4.71 | -11.54 | -9.08 |
| inh6H | 20 | 6.23 | -7.63 | 8.36 | -9.76 | -9.29 |
| inh8 | 17 | 5.29 | -9.6 | 3.79 | -8.1 | -6.41 |
| inh12 | 29 | 9.03 | -5.01 | 9.15 | -5.13 | -7.36 |
Note 1 Entropy, DG, and energy are given in Kcal/mol.
Note 2 Except for inh12, the number of AutoDock runs was 20. Since AutoDock job of a compound with large number of torsions is apt to be endless, the AutoDock run number for inh12 was reduced to 10.
Note 3 Torsional entropy was obtained by multiplying number of torsions by the degree of freedom which is 0.3113 Kcal/mol.
Note 4 DGexperimental was obtained by the equation given in the Background section of the text.
Table 1 shows Gibbs free energies obtained by both the AutoDock3.0 runs and the laboratory experiment for tested inhibitors. The Gibbs free energy of inhibitors obtained by the experiment can be grouped into two classes; the one with lower Gibbs free energies which includes inh1, inh3cis, inh3trans, inh4 and inh6H, and the one with higher Gibbs free energies which includes inh8 and inh12. The Gibbs free energy of inhibitors obtained by the AutoDock3.0 runs can also be grouped into two classes in a similar manner. This indicates that there is a fairly good correlation between the AutoDock3.0 runs and the laboratory experimental results as is stated later in this text .
Figure 3 shows dockings of inh1 and inh8 against HIV protease and the unexploited carbon affinity region (or hydrophobic region) were observed. Thus, additional inhibitors with hydrocarbon side chains extended in that region were created by Sybyl software as inhibitor candidate compounds (shown in Figure 4). Those are the compounds in which various types of hydrocarbon side chains were attached to an amide functional group on the proline ring. The extended side chains for exten0, exten1, exten2, and exten5 consist of saturated alkyl groups, and exten3 and exten4 contain phenyl groups additionally. Those candidate compounds were tested for docking study and the results were shown in Table 2. Exten4 showed lower DG, which was -12.73 Kcal/mol. This was lower than the inh1 DG, which was -11.99 Kcal/mol indicating that exten4 would be a stronger inhibitor of HIV protease. Figure 5 shows the docked mode between exten4 and HIV protease where exten4 was docked fairly superimposingly to inh1. The unexploited carbon affinity region (or hydrophobic region) around the carboxybenzyl (Cbz) side chain was fairly well exploited by the phenyl ring of exten4, and the other unexploited carbon affinity region (or hydrophobic region) around the t-butyl side chain was exploited by the phenyl side chain attached to an amide functional group on the proline ring.
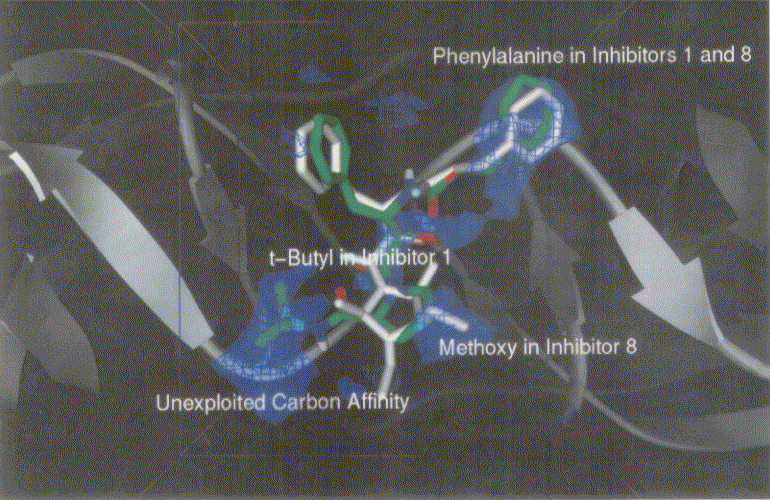
Figure 3. Docking mode of inhibitor 1 (inh1, green) and inhibitor 8 (inh8, white) against HIV protease as obtained by AutoDock3.0. Phenylalanines in inh1 and inh8 are superimposed. Bluish netted areas represent carbon affinity region. Atoms in inh1 are shown by different colors (red, oxygen; blue, nitrogen; and green, carbon).
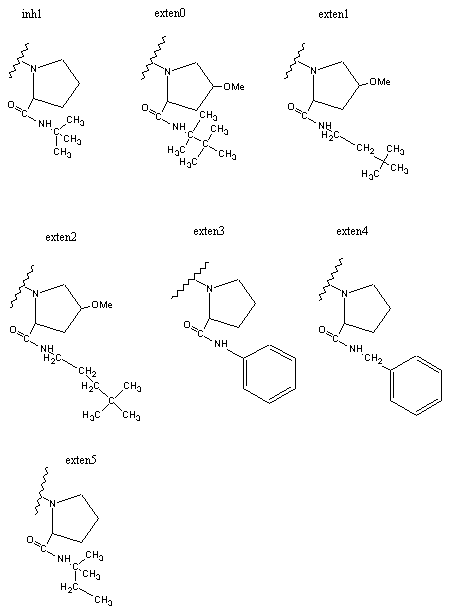
Figure 4. HIV inhibitors tested for AutoDock runs.
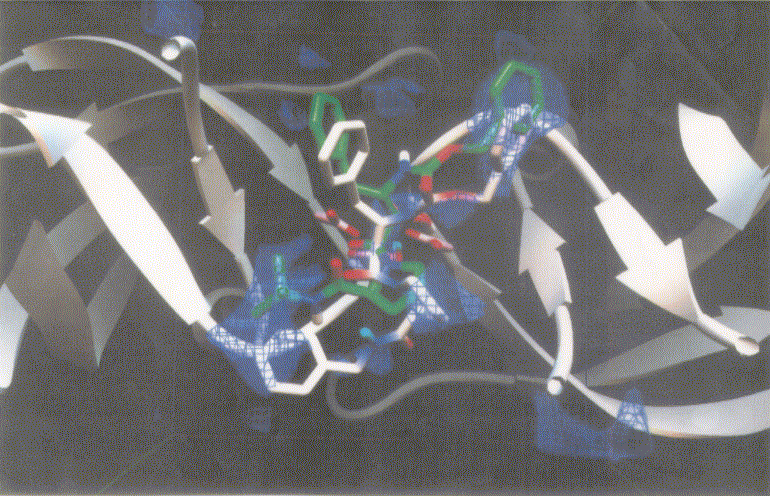
Figure 5. Docking mode of the inhibitor candidate (exten4, white) against HIV protease as obtained by AutoDock3.0. The reference inhibitor 1 (inh1, green) are also shown. Atoms in exten4 and inh1 are showm by different colors (red, oxygen; blue, nitrogen; and white or green, carbon).
Table 2. Gibbs free energy obtained for HIV protease inhibitors predicted by the application of AutoDock3.0 runs.
| inhibitor | number of torsions | torsional entropy (a) | total interaction energy of complex (b) | internal energy of inhibitor (c) | DGAutoDock (a+b-c) |
|---|
| inh1 | 13 | 4.05 | -11.74 | 4.3 | -7.69 |
| exten0 | 14 | 4.36 | -6.66 | 7.75 | -2.3 |
| exten1 | 14 | 4.36 | -11.16 | 4.25 | -6.8 |
| exten2 | 15 | 4.67 | -10.16 | 5.48 | -5.49 |
| exten3 | 13 | 4.05 | -11.74 | 3.33 | -7.69 |
| exten4 | 14 | 4.36 | -14.38 | 2.71 | -7.07 |
| exten5 | 14 | 4.36 | -14.81 | 1.54 | -10.45 |
Note 1 Entropy, DG, and energy are given in Kcal/mol.
Note 2 Torsional entropy was obtained by multiplying number of torsions by the degree of freedom which was 0.3113 Kcal/mol.
Note 3 Structures of inhibitors are shown in Figure 1.
Correlation between Gibbs free energies obtained by AutoDock runs and experimental results were plotted in Figure 6. The straight line was drawn by the least square method. The slope of the line was 0.348, which was a definite positive value and it shows that there is a fairly good correlation between the Gibbs free energies obtained by AutoDock runs and experimental results previously stated. Although the slope value of one would show the best correlation between the two, .the authors believe that the value 0.348 was not so discouraging to state the correlation between the two because of the limitation of comparison between the biological system and computational chemistry due to the fact that there are lots of unsolved areas such as incorporations of the effects of solvation and the protein movements into the system and so on at this time.
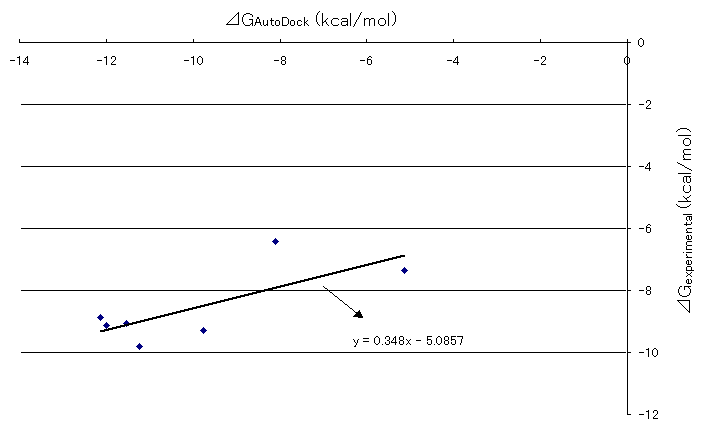
Figure 6. Correlation between AutoDock and experimental Gibb's free energies of HIV and inhibitor docking
5 Conclusions
It was found that compounds inh3trans and inh6H were more powerful inhibitors of HIV protease than the others. Furthermore, there wasn't a big discrepancy between the free energies obtained by AutoDock3.0 and by experimental method. It is feasible to state that the fairly good correlation was found between calculated molecular mechanics interaction energies and experimental inhibitory potencies for a series of HIV protease inhibitors. Based on this favorable correlationship between the computational result and the experimental result, the computational experiment was performed for the Sybyl-drawn potential inhibitors taking into consideration the fact that an unexploited carbon affinity region (or hydrophobic region) with sizable volume was observed in the docking mode for inh1 and inh8 against HIV protease. Those were compounds with t-butyl substituted by hydrophobic side chains to fill the unexploited carbon affinity region. Among those the proposed inhibitor with t-butyl substituted by benzyl group exhibited the lowest docking energy. Since one of the goals of this paper was to perform the computational drug-design experiment to investigate potential HIV protease inhibitors, the authors would like to leave the clinical investigational work for the expertise of those areas.
This work was partly supported by Collaborative Study Grant A of Kobe Gakuin University. This work was also partly supported by a grant for High Technology Research Center from Monbusho, and a Monbusho Grant-in-aid for International Scientific Research.
References
[ 1] Perez, C., Pastor, M., Ortiz, A. R. and Gago, F., Comparative Binding energy analysis of HIV -1 protease inhibitors: incorporation of solvent effects and validation as a powerful tool in receptor-based drug design, J. Med. Chem., 41, 836-852 (1998).
[ 2] Rosin, C. D., Halliday, R. S. and Belew, R. K., A comparison of global and local search methods in drug docking, UCSD CSE Technical Report, #CS97-522, 1-10 (1997).
[ 3] Bohm, H. J., LUDI: rule-based automatic design of new substituents for enzyme inhibitor leads, J. Comput.-Aided Mol Design, 6, 593-606 (1992).
[ 4] Kuntz, I. D., Blaney, J. M., Oatley, S. J., Langride, R. and Ferrin, T. E., A geometric approach to macromolecule-ligand interactions, J Mol Biol, 161, 269-288 (1982).
[ 5] Goodsell, D. S., Lauble, H., Stout, C. D. and Olson, A. J., Automated docking in crystallography : analysis of the substance, Proteins Struc. Funct., 17, 1-10 (1993).
[ 6] Lunney, E. A., Hagen, S. E., Domagala, J. M., Humblet, C., Kosinski, J., Tai, B. D., Wamus, J. S., Wilson, M., Ferguson, D., Hupe, D., Tammio, P. J., Baldwin, E.T., Bhat, T. N. and Liu, B.,N., A novel nonpeptide HIV-1 protease inhibitor: elucidation of the binding mode and its application in the design of related analogs, J. Med. Chem., 37, 2664-2677 (1994).
[ 7] Stoddard, B. L. and Koshland, D.E., Prediction of the structure of a receptor-protein complex using a binary docking method, Nature, 358, 774-776 (1992).
[ 8] Li, M., Morris, G. M., Lee, T., Laco, G. S., Wong, C. H., Olson, A. J., Elder, J. H., Wlodawer, A., and Gustchina, A., Structural studies of FIV and HIV-1 proteases complexed with an efficient inhibitor of FIV protease, Proteins, 38, 29-40 (2000).
[ 9] Beck, A. Q., Hervio, L., Dawson, P. E., Elder, J. H., and Madison, E. L., Identification of efficiently cleaved substrates of JIV-1 protease using a phage display library and use in inhibitor development, Virology, 274, 391-401 (2000).
[10] Bini, E. J., Micale, P. L., and Weinshel, E. H., Natural history of JIV-associated esophageal disease in the era of protease inhibitor therapy, Dig. Dis. Sci., 45, 1301-1307 (2000).
[11] Krzysztof, A., Crystal structures of HIV -1 protease inhibitor complexes, Perspectives in Drug Discovery and Design, 1, 23-48 (1993).
[12] Turner, S. R., Strohbach, J. W., Tommasi, R. A., Aristoff, P. A., Johnson, P. D., Skulnick, H. I., Dolak, L. A., Seest, E. P., Tomich, P. K., Bohanon, M.J., Horng, M. M., Lynn, J. C., Chong, K. T., Hinshaw, R. R., Watenpaugh, K. D., Janakiraman, M. N. and Thaisrivongs, S., Tipranavir (PNU-140690): A potent, orally bioavailable nonpeptidic HIV protease inhibitor of the 5,6-dihydro-4hydroxxy-2-pyrone sulfonamide class, J. Med. Chem., 42, 3467-3476 (1998).
[13] Friedman, S. H., Ganapathi, P. S., Rubin, Y. and Kenyon, G. L., Optimizing the binding of fullenrene inhibitors of the HIV -1 protease through predicted increase in hydrophobic desolvation, J. Med. Chem., 41, 2424-2429 (1998).
[14] Wang, J. M., Ueda, H., Howard, O. M., Grimm, M. C., Chertov, O., Gong, X., Gong, W., Resau, J. H., Broder, C. C., Evan, G., Arthur, L. O., Ruscetti, F. W., and Oppenheim, J. J., HIV-1 envelope gp120 inhibits the monocyte response to chemokines through CD4 signal-dependent chemokine receptor down-regulation, J. Immunol., 161, 4309-4317 (1998).
[15] Chaom, C. C., Gekker, G., Sheng, W. S., Loh, H. H., and Peterson, P. K., Orphan opioid receptor oligonucleotides inhibit HIV-1 expression in human brain cells, Adv. Exp. Med. Biol., 437, 83-90 (1998).
[16] Ueda, H., Howard, O. M., Grimm, M. C., Su, S. B., Gong W., Evans, G., Ruscetti, F. W., Oppenheim, J. J. and Wand, J. M., HIV-1 envelope gp41 is a potent inhibitor of chemoattractant receptor expression and function in monocytes, J. Clin. Invest., 102, 804-812 (1998).
[17] Morris, G. M., Goodsell, D. S., Halliday, R.S., Huey, R., Hart, W. E., Belew, R. K. and Olson, A. J., Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function, J. Computational Chemistry, 19, 1639-1662 (1998).
[18] Lee, T., Le, V.-D., Lim, D., Lin, Y.-C., Morris, G.M., Wong, A.L., Olson, A.J., Elder, J.H. and Wong, C.-H., Development of a new type of protease inhibitors, efficacious against FIV and HIV variants, J. Am. Chem. Soc., 121, 1145-1155 (1999).
Return