Automated Identification of Three-Dimensional Common Structural Features of Proteins
Hiroaki KATO and Yoshimasa TAKAHASHI
 Return
Return
1 Introduction
Structural feature analysis and similarity analysis of proteins can give us a lot of useful information for molecular biological science and related areas. With the rapidly increasing number of proteins whose three-dimensional (3D) structures are known, efficient approaches are required for a systematic analysis of the 3D structural feature of proteins. The identification of 3D structural similarity presents an enormous computational challenge. There are a number of computational methods, which one could use to estimate, in a statistical manner, the similarity between proteins [1 - 3]. Alternatively, there are also approaches that search for superimposable fragment at the backbone coordinate level [4 - 6] and the secondary structure level [7 - 9]. Particularly, Mitchell et al. [10] describe a sophisticated computer program, called POSSUM, that allows searching for a secondary structure motif in a protein structure database. The program was based on a substructure search algorithm for ordinary organic molecules. Their work has been extended by Grindley et al. [11] for 3D motif finding, but not for substructure searching with a query pattern. In our preceding works, we have also investigated the approaches to the 3D substructure search or motif search of proteins [12, 13]. A computer program, called SS3D-P, has been developed for 3D substructure search of proteins [12]. SS3D-P allows us to identify all occurrences of a 3D query pattern (or a 3D motif) consisting not only of chain-based peptide segments (e.g. P-loop motif) but also of a set of disconnected amino acid residues (e.g. two cysteine residues and two histidine residues of Zinc-finger motif) for the protein structure databases. The authors also reported another program called SS3D-P2 for a 3D structure motif search of proteins based on the secondary structure elements (SSEs), a-helices and b-strands [13]. In this paper, we extend these studies into and describe a computer program called AIM (Automated Identification of three-dimensional Motif of proteins) which can be useful for finding new motif candidates or 3D common structural features of proteins.
2 Methods
In the present work, to avoid the need to consider the thousands of atoms of proteins, the Ca approximations have been adopted for the identification of 3D patterns of amino acid residues that constitute a particular spatial arrangement of certain types of secondary structure. The identification of individual secondary structure segments was carried out using Kabsch and Sander's method [14]. In this manner, a-helix and b-strand secondary structure segments are described with vectors in 3D space. Further reduction of the structure representation was employed, in which each secondary structure segment described with a vector is reduced into a conceptual point that is labeled with starting and ending residues, the length of the segment (the distance between the two residues) and the type of secondary structure. Thus, the whole structure of a protein can be represented by a set of the conceptual points that involve only the secondary structure segments identified within the protein. This set of points can be regarded as a graph in mathematical graph theory. The approximation described above allows us to highly reduce a protein structure which consists of thousands atoms or points in 3D space. A maximal common subgraph matching algorithm based on the graph theoretical clique finding procedure [15] was used for the search for the geometrical patterns which are common in the proteins under investigation. The basic procedure consists of two major parts, (1) generation of the docking graph from two protein molecular graphs obtained by the method just mentioned, and (2) clique finding for the docking graph. These processes are similar to those of our previous work [13] except for the higher reduced representation of protein molecular graphs and the docking graph derived from the graphs. However, the present approach doesn't require any query pattern to search for. The matter is very different from the previous work. No query pattern is used and no particular peptide sequence segment is assumed to be involved. Three-dimensional molecular coordinates of proteins are only required in this work. This means that we have to still handle a large docking graph (in terms of the number of vertices) to find the common geometrical features between two or more protein molecules even when we use the SSE representation mentioned above. To reduce the sign of the docking graph to consider and to reduce the computational requirement to find the clique within the graph, a higher reduced representation of protein molecular graph and an alternative docking procedure with it are introduced here.
Figure 1 shows an illustrative example for the highly reduced graph representation of protein molecules and a docking graph derived from them. In this work, the docking graph Doc(Ga', Gb') for graph Ga' and Gb' in Figure 1 was derived from the following conditions;
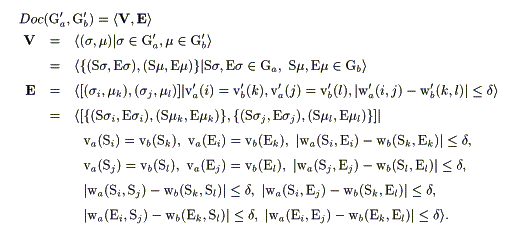
In the equations, V and E are the set of vertices and the set of edges of the docking graph respectively. s stands for a vertex element from the graph Ga' and m stands for the one that comes from Gb' to be compared. Ss and Es stand for the starting and ending residues of a secondary structure element s in Ga' respectively. Sm and Em stand for those of the segment m. va'(i) stands for the weight on the i-th vertex of Ga' which involves a pair of the starting residue and the ending residue, the length (i.e. the distance between the two residues) and the type of SSE for the i-th segment of protein a. vb'(k) represents the weight on the k-th vertex of Gb' in the same manner. wa'(i, j) stands for the weight on the edge that joins the i-th and the j-th vertices in Ga', which involves the spatial arrangement in the 3D molecular space. wb'(k, l) stands for that of Gb'. On the other hand, va(Si) stands for the weight on the vertex Si in the protein molecular graph Ga that involves the type of SSE and the starting residue. vb(El) stands for that on the vertex El in Gb, which involves the type of SSE and the ending residue. wa(Si, Ei) stands for the weight on the edge that joins Si and Ei, which involves the length of the SSE i of the protein a. wa(Si, Sj) represents the weight on the edge that joins Si, and Sj, which involves the distance between the starting residue of SSE i and that of SSE j in protein a. wa(Ei, Ej) stands for that on the edge between the ending residues. Others are in the similar manner. Here, d is a tolerance (A) specified at the searching. As described above, in the present work, a directed graph representation was used to represent the sequence direction of peptide segments of SSEs in describing the protein molecular graphs to be compared. It allows us to distinguish different sequence directions (N-terminal to C-terminal, or C-terminal to N-terminal) of the SSEs if we want to do this at the searching. All the processes were fully computerized and implemented into a computer program, AIM. For the clique finding, a tree-search method based on the backtracking procedure was used. As the basic algorithm for the clique search used here was reported elsewhere [16], the description will not be repeated now. The whole program was written in the standard C language, and all of the computational works in the current paper were carried out on Silicon Graphics Inc. Indy workstation with IRIX operation system.
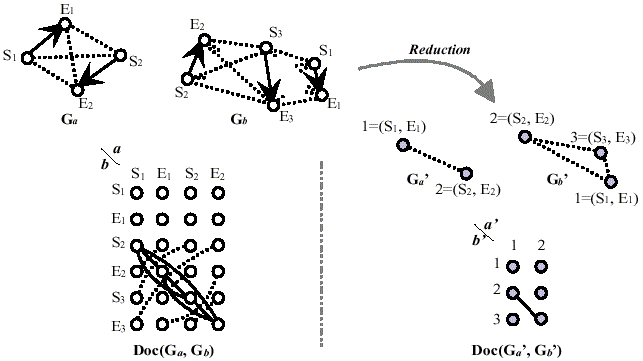
Figure 1. Highly reduced graph representation of three-dimensional structure of proteins and a docking graph. Ga and Gb stand for molecular graphs of proteins. Si and Ei are the starting residue and the ending residue of i-th secondary structure element in each protein. Doc(Ga, Gb) is a docking graph for graph Ga and Gb. Ga' and Gb' stand for further reduced graphs of Ga and Gb. (Si, Ei) is a label (or weight) for the reduced vertex that involves i-th secondary structure element of a protein molecular graph. Figures (1 and 2 in Ga', and 1, 2 and 3 in Gb') stand for their new vertex identifiers. Doc(Ga', Gb') is a new docking graph from further reduced protein molecular graphs Ga' and Gb'.
3 Results and discussion
To test the performance of our program AIM, alcohol dehydrogenase (1DHXA) and lactate dehydrogenase (9LDTA) were used for a trial of the automated identification of 3D common structural features. It is known that they are NAD-dependent dehydrogenases and have typical Rossmann-fold motif [17]. The 3D coordinate data were taken from the PDB (Protein Data Bank) file [18] and were represented using the reduced representation described above. The trial was carried out using a searching condition in which the different kinds of secondary structure segments were distinguished, the directions of each secondary structure segment on the primary sequence were considered, and the tolerance value of the distance was set to be 5.0A. For these two proteins, the AIM identified five distinct patterns as the common structural features that are maximal in terms of the number of SSEs. The results obtained are listed in Table 1. On the fifth pattern in Table 1, six parallel b-strands within 1HDXA (T194-F198, R218-V222, E239-I241, F264-E267, T288-I291 and T313-G316) and those within 9LDTA (L93-I96, K134-V137, V160-G162, K23-V27, E48-V52 and K77-G81) were successfully identified as one of the maximal 3D common substructures of these proteins at the SSE representation level. The graphical views of the 3D structures of these proteins and the detected segments are displayed in Figure 2. These sites correspond to the six parallel strand segments that form a Rossmann-fold motif known as an NAD-binding domain [17]. The finding results often depend on the tolerance value d, a small value of the tolerance may find well-defined 3D common structural moieties of the proteins. However, it may lead us to miss finding for some of other meaningful features that may be found with some more loose restriction in the 3D special arrangement. For comparison, we also carried out a trial with the value of 7.0A for the tolerance at the search. In this case, the AIM identified a larger set of secondary structure segments that involved three a-helix segments adding to the six parallel b-strand segments. Those segments identified are listed in Table 2. For the second pattern in Table 2, the graphical views of their corresponding sites on each protein are shown in Figure 3. It is again apparent that the site corresponds more closely to the Rossmann-fold motif. The result shows that the program successfully identified the motif candidate as a 3D common structural feature of these proteins.
Table 1. Three-dimensional common structural features between alcohol dehydrogenase (1DHXA) and lactate dehydrogenase (9LDTA) found by AIM.
| ID No. | PDB | Identified site (amino acid identifier) |
|---|
| NO. 1 |
| [ 1_01] | 1HDXA | R101 K104 T194 F198 G202 A213 R218 V222 F264 E267 T313 G316 |
| [ 2_01] | 9LDTA | W203 G205 K 23 V 27 A 31 M 42 E 48 V 52 L 93 I 96 V160 G162 |
| NO. 2 |
| [ 1_01] | 1HDXA | T194 F198 G202 A213 R218 V222 F264 E267 T288 I291 T313 G316 |
| [ 2_01] | 9LDTA | K 23 V 27 A 31 M 42 E 48 V 52 L 93 I 96 K134 V137 V160 G162 |
| [ 02] | 9LDTA | E 48 V 52 A 31 M 42 K 77 G 81 K 23 V 27 L 93 I 96 K134 V137 |
| NO. 3 |
| [ 1_01] | 1HDXA | T194 F198 G202 A213 R218 V222 E239 I241 F264 E267 T288 I291 |
| [ 2_01] | 9LDTA | K 23 V 27 A 31 M 42 L 93 I 96 K134 V137 E 48 V 52 K 77 G 81 |
| NO. 4 |
| [ 1_01] | 1HDXA | T194 F198 R218 V222 E239 I241 F264 E267 T288 I291 M306 L309 |
| [ 2_01] | 9LDTA | L 93 I 96 K134 V137 V160 G162 K 23 V 27 E 48 V 52 S 70 F 72 |
| NO. 5 |
| [ 1_01] | 1HDXA | T194 F198 R218 V222 E239 I241 F264 E267 T288 I291 T313 G316 |
| [ 2_01] | 9LDTA | L 93 I 96 K134 V137 V160 G162 K 23 V 27 E 48 V 52 K 77 G 81 |
The trial was carried out under the following conditions: the tolerance value of the distance was 5.0A, the directions of each secondary structure segment on the primary sequence were considered. Each secondary structure segment on the identified site is shown with a pair of amino acid identifiers of the N-terminal and the C-terminal side.
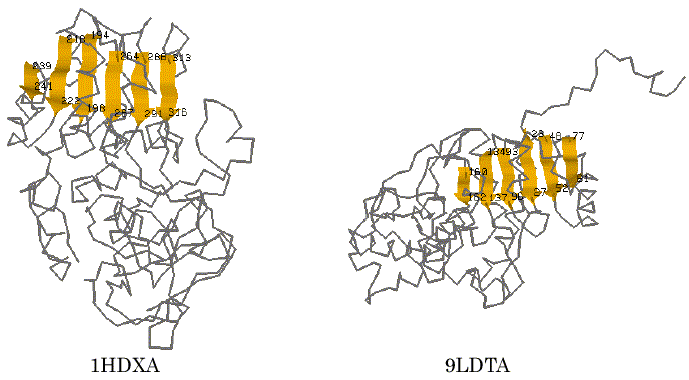
Figure 2. Graphical views of the 3D common structural features found by AIM. The visualization is for No.5 in Table 1. Peptide segments identified are in color.
Table 2. Three-dimensional common structural features between alcohol dehydrogenase (1DHXA) and lactate dehydrogenase (9LDTA) found by AIM with the tolerance of 7.0 A.
| ID No. | PDB | Identified site (amino acid identifier) |
|---|
| NO. 1 |
| [ 1_01] | 1HDXA | R101 K104 T194 F198 G202 A213 R218 V222 E239 I241 I250 M257 F264 E267 T288 I291 T313 G316 |
| [ 2_01] | 9LDTA | W203 G205 K 23 V 27 A 31 M 42 E 48 V 52 K 77 G 81 Y 85 T 88 L 93 I 96 K134 V137 V160 G162 |
| NO. 2 |
| [ 1_01] | 1HDXA | T194 F198 R218 V222 F229 E234 E239 I241 F264 E267 L272 L280 T288 I291 M306 L309 T313 G316 |
| [ 2_01] | 9LDTA | L 93 I 96 K134 V137 V142 I152 V160 G162 K 23 V 27 A 31 M 42 E 48 V 52 S 70 F 72 K 77 G 81 |
The trial was carried out under the same conditions excepting the tolerance value.
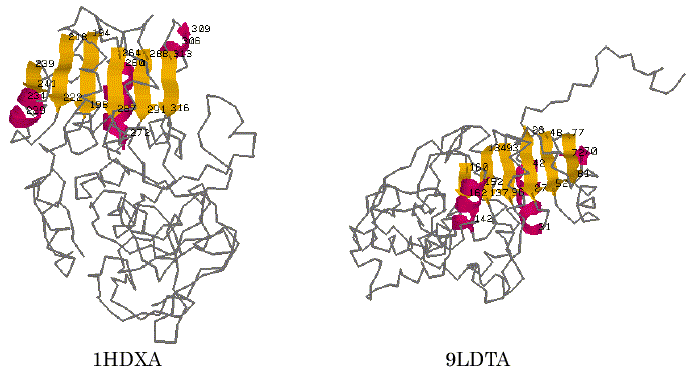
Figure 3. Graphical views of the 3D common structural features found by AIM. The visualization is for the result of No.2 in Table 2.
Subsequently, we also tried a 3D substructure search with a query of the 3D structural feature obtained above. The substructure searching was carried out by the use of a 3D substructure search program, SS3D-P2, which was developed in our previous work [13]. The database that contains 521 proteins taken from the PDB file was prepared and used for this computational trial. The 3D query pattern used in this trial was based on that of the six segments of parallel b-strands identified by AIM for 1HDXA. The searching result with the tolerance of 6.0A is shown in Table 3. In addition to 1HDXA and 9LDTA, the program successfully found the corresponding sites in d-glycerate dehydrogenase (1GDHA) and malate dehydrogenase (2CMD) and others that are known to have the Rossmann-fold motif excepting 3-phosphoglycerate kinase (1PHP) and periplasmic binding protein (2LIV) in SCOP database [19]. The latter two proteins, both 1PHP and 2LIV, are not categorized into a superfamily that has NAD(P)-binding Rossmann-fold domains, but it was validated in SCOP database searching that both of them also have the typical parallel beta-sheet of 6 strands with different sequence topology. Some of the graphical views of the hit proteins and the sites identified within them are shown in Figure 4. The result shows that the AIM is successfully applicable to finding new candidates of 3D structure motif of proteins.
Table 3. Results of the 3D substructure search for 521 proteins by SS3D-P2.
| ID No. | PDB | Protein | Hit site (amino acid identifier) |
|---|
| [ 131_01] | 1GDHA | D-GLYCERATE DEHYDROGENASE | T149 Y153 D172 F176 T192 F193 F206 L209 A233 N237 L258 F263 | + |
| [ 156_01] | 1HDXA | ALCOHOL DEHYDROGENASE | T194 F198 R218 V222 E239 I241 F264 E267 T288 I291 T313 G316 | + |
| [ 195_01] | 1LDM | LACTATE DEHYDROGENASE | K 22 V 26 E 47 V 51 K 76 S 79 L 91 I 94 I132 V135 I158 G160 | + |
| [ 196_01] | 1LDNA | L-LACTATE DEHYDROGENASE | R 22 I 26 E 47 I 51 D 77 H 80 L 91 I 94 L132 V135 V158 G160 | + |
| [ 205_01] | 1LLDA | L-LACTATE DEHYDROGENASE | M 78 I 81 I119 L122 I145 G147 K 9 I 13 E 34 E 38 S 63 S 67 | + |
| [ 265_01] | 1PHP | 3-PHOSPHOGLYCERATE KINASE | F189 I193 Y345 I348 H368 I369 L312 N316 N214 I217 R259 Y261 | |
| [ 406_01] | 2CMD | MALATE DEHYDROGENASE | V 72 I 75 C113 I116 L143 G145 K 2 L 6 E 29 Y 33 K 54 F 58 | + |
| [ 442_01] | 2LIV | PERIPLASMIC BINDING PROTEIN | Q221 P225 L245 K248 A340 D342 F196 G200 I141 H145 V169 G174 | |
| [ 456_01] | 2OHXA | ALCOHOL DEHYDROGENASE | T194 F198 R218 V222 E239 V241 F264 E267 V288 I291 T313 G316 | + |
| [ 578_01] | 9LDTA | LACTATE DEHYDROGENASE | L 93 I 96 K134 V137 V160 G162 K 23 V 27 E 48 V 52 K 77 G 81 | + |
The search was carried out under the following conditions: the tolerance value of the distance was 6.0A, the directions of each secondary structure segment on the primary sequence were considered. The symbol '+' in the last column shows the proteins that are known in the SCOP database.
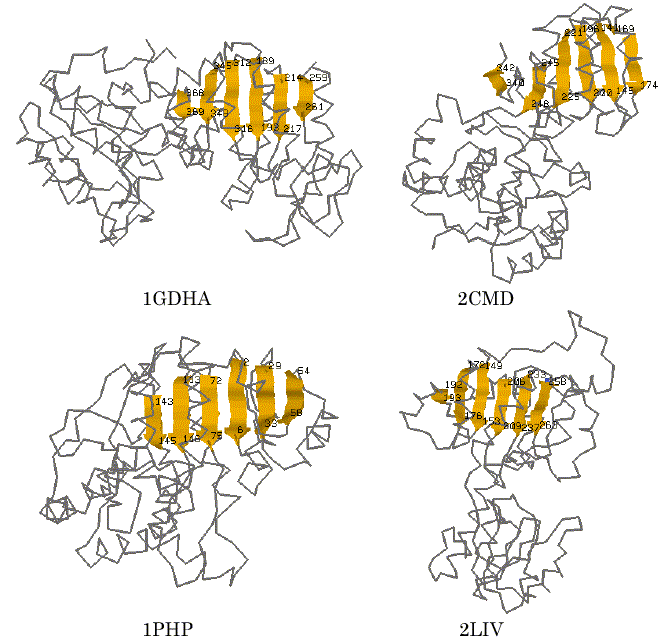
Figure 4. Visualization of the detected sites of the proteins hit by 3D substructure searching for the segments similar to the six parallel b-strands of 1HDXA identified by AIM.
Obviously, the AIM doesn't require any query pattern to search for in advance. Three-dimensional molecular coordinates of proteins are only required in this work. Once reasonable three-dimensional molecular coordinates of proteins have been obtained, the AIM can perform automated search operations to provide common geometrical structural features among them that may be candidate patterns of three-dimensional structural motif. The results given in this paper show that the present approach is successfully applicable for finding new candidates of the 3D motif of proteins.
This work was supported by a Grant-in-Aid for Scientific Research on Priority Areas 'Genome Science', from The Ministry of Education, Science, Sports and Culture of Japan. We used computer resources at the Human Genome Center, Institute of Medical Science, The University of Tokyo.
References
[ 1] Remington, S.J. and Matthews, B.W., Proc. Natl. Acad. Sci. USA, 75, 2180 (1978).
[ 2] Karpen, M.E., deHaseth, P.L. and Neet, K.E., Proteins, 6, 155 (1989).
[ 3] Lessel, U. and Schomburg D., Protein Eng., 7, 1175 (1994).
[ 4] Taylor, W.R. and Orengo, C.A., J. Mol. Biol., 208, 1 (1989).
[ 5] Vriend, G. and Sander, C., Proteins, 11, 52 (1991).
[ 6] Alexandrov, N.N., Takahashi, K. and Go, N., J. Mol. Biol., 225, 5 (1992).
[ 7] Abagyan, R. A. and Maiorov, V. N., J. Biomol. Struct. Dynam., 6, 1045 (1989).
[ 8] Mizuguchi, K. and Go, N., Protein Eng., 8, 335 (1995).
[ 9] Holm, L. and Sander, C., Intelli. Sys. Mol. Biol., 3, 179 (1995).
[10] Mitchell, E. M., Artymiuk, P. J., Rice, D. W. and Willett, P., J. Mol. Biol., 212, 151 (1989).
[11] Grindley, H. M., Artymiuk, P. J., Rice, D. W. and Willett, P., J. Mol. Biol., 229, 707 (1993).
[12] Kato, H. and Takahashi, Y., Bull. Chem. Soc. Jpn., 70, 1523 (1997).
[13] Kato, H. and Takahashi, Y., Comput. Applic. Biosci., 13, 593 (1997).
[14] Kabsch, W. and Sander, C., Biopolymers, 22, 2577 (1983).
[15] Bron, C. and Kerbosh, J., Commun. ACM, 16, 575 (1973).
[16] Takahashi, Y., Maeda, S. and Sasaki, S., Anal. Chim. Acta, 200, 363 (1987).
[17] Rossmann, M.G., Moras, D. and Olsen, K.W., Nature, 250, 194 (1974).
[18] Bernstein, F.C., Koetzle, T.F., Williams, G.J.B., Meyer,Jr, E.F., Brice, M.D., Rodgers, J. R., Kennard, O., Shimanouchi T. and Tasumi, M., J. Mol. Biol., 112, 535 (1977).
[19] Murzin, A.G. Brenner, S.E., Hubbard, T. and Chothia, C., J. Mol. Biol., 247, 536 (1995).
Return