Development of a Program for Construction of
a Starting Material Library for AIPHOS
Koji SATOH, Shukou AZUMA, Hiroko SATOH1) and Kimito FUNATSU
 Return
Return
Introduction
Since the seminal publication on computer-assisted organic synthesis by Corey and Wipke2) was presented, more than fifty computer-assisted synthesis planning systems, as we know, for designing syntheses of organic compounds have been developed during the last twenty five years. There are two categories in computer-assisted organic synthesis planning systems. One is an empirical knowledge-based approach, such as in LHASA3) or SECS.4) The other is a logic-centered approach, such as in SYNGEN5) or EROS.6) The advantage with regard to the former systems is that their proposals of retrosyntheses can generally be executed in laboratories. The latter ones have the merit that their proposed retorosynthetic paths involve original and novel ones. Both two approaches are attractive for chemists.
AIPHOS (Artificial Intelligence for Planning and Handling Organic Synthesis),7) which has been developed in our laboratory, combines the merits of the empirical knowledge-based approach and the logic-centered approach. That is, AIPHOS can propose original and new retrosynthetic routes to assure their implementation in laboratories because of its unique reaction knowledge base.8) For the moment, the AIPHOS system proceeds stepwise, interactively, determining at each step the synthetic precursors from molecules of the former step. The synthesis design procedure of AIPHOS is shown in Figure 1. A series of procedures in the current AIPHOS is as follows: (1) Input of a desired target structure is made. (2) Plausible strategic sites in the target molecule are obtained. In acquiring strategic sites, the topological strategy and the functional group based strategy which are described by Corey9) are applied to the target molecule. One of the proposed strategic sites is selected by the user. When the user wishes to specify strategic sites according to his/her synthetic strategies, he/she may edit his/her own strategic sites manually. (3) Possible precursors on the basis of the strategic sites selected at step 2 are generated. One set of proposed precursors is selected by the user. (4) Appropriate leaving groups are added to a set of precursors selected at step 3 automatically by utilizing the leaving groups knowledge base. If necessary, the user may add leaving groups manually. (5) The proposed retrosynthetic path is evaluated as to whether it can occur or not with the reaction knowledge base. If the proposed retrosynthetic path can occur, it is displayed to the user with related reaction schemes in the AIPHOS database.
In planning of organic syntheses, chemists look for efficient retrosyntheses analyzing a relationship between a proper unit of the target molecule and the structure of available compounds like starting materials offered in catalogs. Other synthesis planning systems such as LHASA,10) SECS11) and SYNCHEM12) embody this kind of analysis of chemists by using their original starting material libraries approaches to starting material-based retrosynthetic analysis. In order to introduce a similar approach to AIPHOS, we have developed an efficient program which can incorporate data of a database of chemical structures on the market into the library. This program is also able to recognize synthetic equivalents to be categorized functional groups. And then, the resulting library is made up of a hierarchical structure using abstract graphs. In this paper, we report a structure, a construction method of our original starting material library and the registration results of data selected at random from a commercial chemical structure database. The starting material-oriented retrosynthetic analysis in AIPHOS will be reported in a future paper.13)
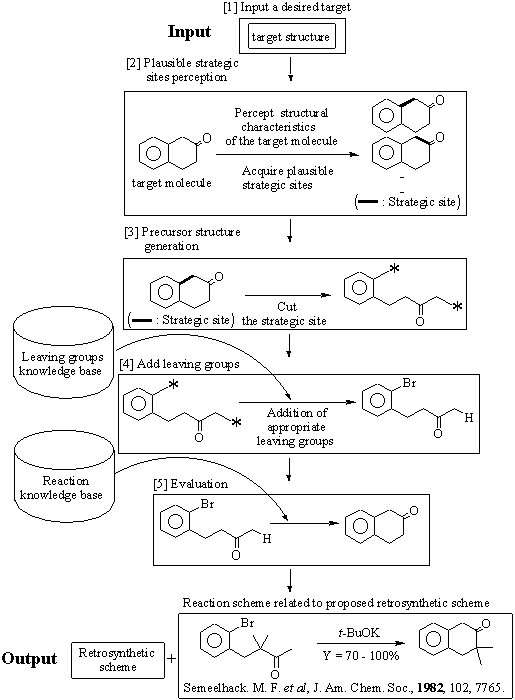
Figure 1. Block diagram of AIPHOS
Abstract Graphs
A hierarchy of abstract search spaces similar to Sacerdoti's hierarchical planning14) is selected for building the starting material library. The main reason it was chosen is that hierarchy ignores irrelevant connections, the number of compared graphs becomes smaller than that of original graphs for abstraction, and the time required registration of data can be shortened.
Four levels of abstract structures are used. (1) The S. M. Data level consists of completely specified starting materials. (2) The low level contains information about positions and kinds of functional groups recognized according to functional group codes. (3) The high level, in which functional groups are replaced with X, contains only information about their positions. (4) The top level comprises basic skeletons of starting materials. If nonaromatic C-C multiple bonds exist in abstract graphs, they are converted into single bonds at this level. In Figure 2, an example of abstract graphs at each level is shown.
Recognition of Positions and Kinds of Functional Groups
One of the characteristic points of our starting material library lies in exact recognition of positions and kinds of functional groups. For this recognition, we make use of 111 structural characteristic keys which feature reaction sites and their environments in AIPHOS. Some of them are listed in Table 1. The concept of how to establish them is described in a previous paper.15)
In principle, abstract graphs must contain basic skeletons of compounds in building the starting material library. Hence, only a part of the above keys, which represent functional groups, is used. Further, several new structural characteristic keys (e.g.; -NHAc, -NHCO2R) are added on because many N-substituted compounds are commercially available as starting materials for various compounds.
Categorization of Functional Groups
When an efficient starting material library is constructed, reducing spaces for abstract spaces is important. Therefore, the concept of synthetic equivalents, which are afforded only for changing functional groups, is applied for abstraction of data. Forty functional groups are categorized into 9 groups shown in Figure 3. If necessary, more functional groups can be additionally registered and the combination of the functional groups in the categories also can be changed by users/chemists.
Abstraction of Starting Materials
An example (benzoic acid), for explanation of abstraction, is given in Figure 4, because it is simple to explain. -CO2H is recognized as a functional group. The structural characteristic key no. is 36, and node number 4 is assigned as a root node of the functional group in recognition of structural characteristic key.
Abstract graphs are derived as follows; The core node comprising the functional group characterized above is replaced with X7, which is the functional group code corresponding to -CO2H. The low level abstract graph is prepared by deleting nodes consisting of the functional group. The high level abstract graph is made by changing X7 in the low level abstract graph to X. The top level abstract graph is given by deleting X in the high level abstract graph (Figure 5). Esters, hetero mixed ethers (e.g.; R1-O-R2, R1-S-R2, R1-NH-R2) and carbamates only consisting of their alkyl groups (methyl, ethyl, i-propyl, t-butyl) are abstracted, because they are frequently employed as starting materials in organic synthesis. The above functional groups consisting of other alkyl groups are not abstracted, and compounds in which any structural characteristics are not recognized are also registered without abstraction.
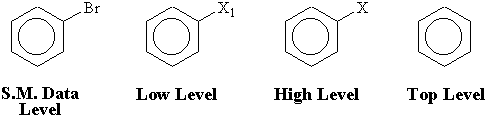
Figure 2. Examples of abstract graphs. X1 is a code arranging functional groups.
X is converted from functional groups codes.
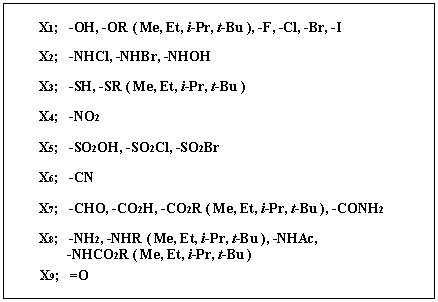
Figure 3. Categories of functional groups.
Table 1. Some structural characteristic keys in the reaction knowledge base in AIPHOS.
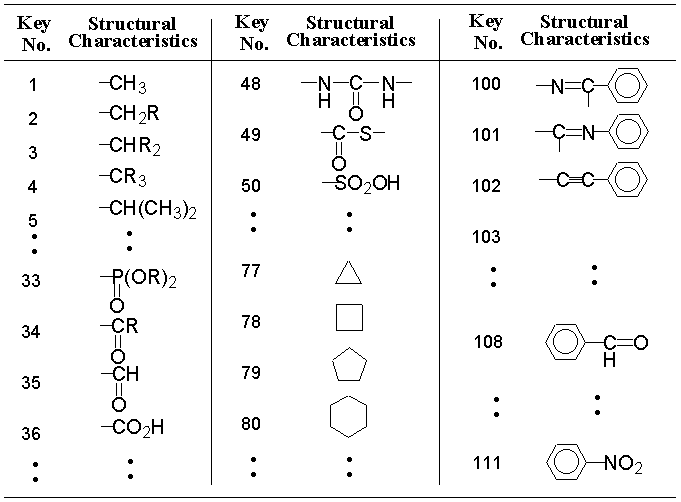
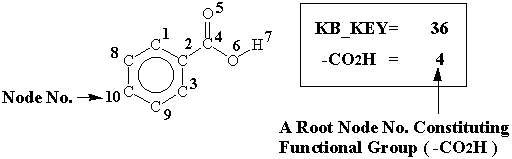
Figure 4. Recognition of structural characteristics in the case of benzoic acid
Decision of Identification
A single compound must not be registered with duplication in constructing the starting material library. It is necessary to register only new compounds. At this time, a Set Reduction algorithm16) for detecting the existence of the same chemical structure is taken. As a Set Reduction algorithm is essentially used for substructure search, a new registering compound and a compound already registered in the library are cross referred by the Set Reduction method for finding structural identification.
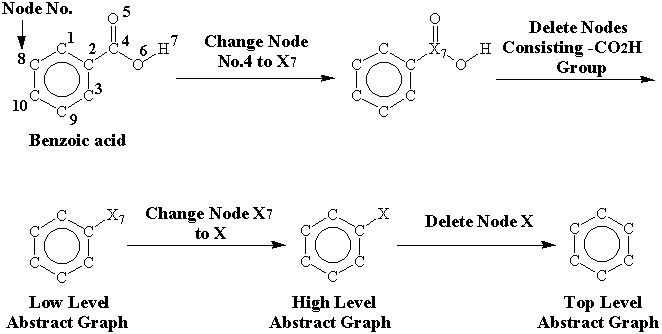
Figure 5. An example of abstraction on benzoic acid
Structure of Starting Material Library
The structure of the starting material library is separated into three blocks: i.e. chiral pool, aromatic pool, nonaromatic pool. In particular, a chiral pool is suited to precursors related to an optically active target molecule. Furthermore, each pool is separated according to the number of carbon atoms contained in top level abstract graphs.
Construction of the AIPHOS Starting Material Library
The starting material library for our purpose requires thousands of data. As it is inefficient to register such a large amount of data manually, the Available Chemicals Directory (ACD)17) containing approximately 130,000 entries of fine chemicals and bulks is introduced into the library. The data having aromaticity in ACD are described by nonaromatic bond information, but aromatic parts of precursors exported from AIPHOS have aromatic bond information. If one chemical structure is described in a different manner (e.g.; bond information), the structure is recognized as a different structure by the Set Reduction method. For solving this problem, before registering data of ACD into the library, the bond information of data having aromaticity is converted to aromatic bond information using the aromaticity recognition program18) based on the Huckel rule. Although our final purpose is to build a large starting material library, five thousand data, which had been selected from ACD at random, were used, to begin with. The numbers of carbon atoms in the materials are from three to ten, since organic chemists use them as starting materials in many cases. The result is shown in Table 2. The percentage of automatically registered data from ACD was 94%. The main factor which did not give 100% registration is that our processing discarded salts such as hydrochloride, potassium and sodium. The percentage of reduction from S.M. data level to top level was 46%. This might be a satisfying result, considering that data were selected at random from ACD.
The flow chart for constructing the starting material library is diagrammed in Fig. 6.
Table 2. A result of registration from ACD.

Conclusions
We have developed a program for constructing a fundamental starting material library to use starting material-oriented retrosynthetic analysis in AIPHOS. This program can import data of other commercial chemical structure databases as well as data of ACD into the library, if their structural data are described by MDL mole file format or can be converted to its file format. Synthetic equivalents are also recognized by using this program. The resultant library is made up of a hierarchical structure using abstract graphs. This library could be used to decide which step in retrosynthetic routes proposed by AIPHOS should be terminated.
Now, further investigations for registering much more data, such as all data in ACD, in the library automatically are in progress.
Experimental
In the starting material library, we can deal with the following atom types (C, H, N, O, P, S, F, Cl, Br, or I) and less than 128 atoms. The program is written in FORTRAN 77. The program is currently running on a SGI-INDY system (R4400, 200MHz).
Acknowledgment
We thank Dr. Ryutaro Kishimoto and Molecular Design, Ltd., for permission to use data from the Available Chemicals Directory and also acknowledge Mrs. Atomi Yoshida and Mr. Yasuhiko Yotsui of the Information System Department, Daiichi Pharmaceutical Co. Ltd., for assisting in those preparations.
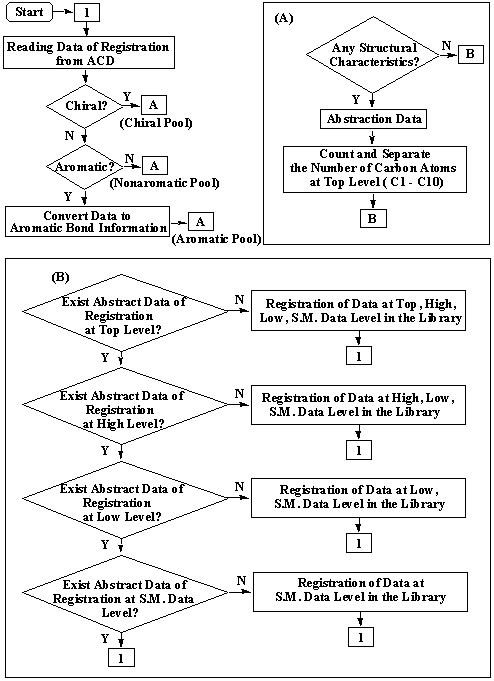
Figure 6. A flow chart for construction of a starting material library.
References and notes
(1) Present Address: Synthetic Organic Chemistry Laboratory, The Institute of Physical and Chemical Research (RIKEN), Wako, Saitama, 351-01 Japan.
(2) E. J. Corey and W. T. Wipke, Science, 166, 178 (1969).
(3) D. A. Pensak and E. J. Corey, "Computer-Assisted Organic Synthesis", American
Chemical Society, Washington, DC (1978), pp. 1-32.
(4) W. T. Wipke, G. I. Ouchi and S. Krishnan, Artif. Intell., 11, 173 (1978).
(5) J. B. Hendrickson, E. Braun-Keller and G. A. Toczko, Tetrahedron, 37, 359 (1981).
(6) J. Gasteiger, Chim. Ind. (Milan), 64, 714 (1982).
(7) K. Funatsu and S. Sasaki, Tetrahedron Comput. Method., 1, 27 (1988).
(8) H. Satoh and K. Funatsu, J. Chem. Inf. Comput. Sci., 35, 34 (1995).
(9) E. J. Corey, Angew. Chem. Int. Ed. Engl., 30, 455 (1991).
(10) A. P. Johnson, C. Marshall and P. N. Judson, Rec. Trav. Chim. Pays-Bas., 111, 310(1992).
(11) W. T. Wipke and D. Rogers, J. Chem. Inf. Comput. Sci., 24, 71 (1982).
(12) H. Gelernter, A. Sanders, D. L. Larson, K. K. Agarwal, R. H. Boivie, G. A. Spritzner, and J. E. Searlman, Science, 197, 1041 (1977).
(13) K. Funatsu, T. Yoshino and K. Satoh, in preparation.
(14) E. D. Sacerdoti, Artif. Intell., 5, 115 (1974).
(15) K. Funatsu, C. A. Del Carpio and S. Sasaki, Tetrahedron Comput. Method. 1, 39 (1988).
(16) E. H. Sussenguth, J. Chem. Doc., 5, 36 (1963).
(17) The Available Chemicals Directory is supplied by Molecular Design Ltd., 2132 Farallon Drive, San Leando, CA 94577, United States of America.
(18) B. L. Rose-Kozel and W. L. Jorgensen, J. Chem. Inf. Comput. Sci., 21, 204 (1981).
Return