Estimation of the Redundancy in Human Genome Shotgun Sequencing by a Monte-Carlo Simulation
Ayumu SAITO and Koichi NISHIGAKI
 Return
Return
1 Introduction
Human genome project is now confronted with the crucial problem whether a whole genome shotgun method, which has worked for sequencing bacterial genomes, is still effective to sequencing the human genome, which is thousands times larger than those of bacteria [1 - 3]. There have been currently strong objections to applying a whole genome shotgun method to human genome sequencing mainly because of the obscurity in its efficiency, besides the reason that massive projects based on the conventional strategies have been functioning [2 - 4]. Formerly, Lander and Waterman, who had mathematically worked out with a shotgun process, suggested the effectiveness of shotgun process for genome sequencing and recommended a computer simulation for further discussion [5]. Recently, Weber and Myers published a strategical paper which strongly supports the whole genome shotgun sequencing of human genome [6]. The central problem of the above dispute can be assigned to be what is the redundancy in the shotgun sequencing of human genome. In such a context, this paper aims to give a scientific solution for this problem with the use of Monte-Carlo simulation.
2 Method
A computer program ( Yuki-com2) was worked out using Visual Basic Version 5, and operated under the environment of Microsoft Windows NT4.0 installed on an IBM DOS/V compatible computer [Briefly, mother board, S1682 (Tahoe2ATX); CPU Pentium II 266MHz×2; 32Mbytes EDO-RAM×4; and HDD, E-IDE 4.3 Gbytes×2] assembled at our laboratory from a commercially supplied kit and separately-obtained units.
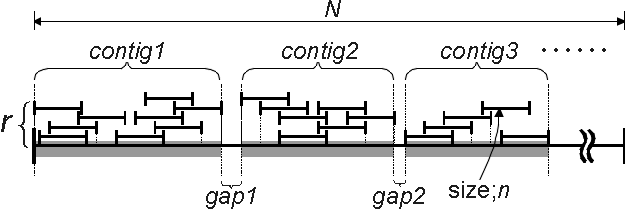
Figure 1. Parameters and variables used for the Monte-Carlo simulation. A genome sequence of N nucleotides in length was allotted to an array, A(i), of N elements on computer. A piece of snow (i.e., a sequence of n nucleotides) was subjected to fall on a site which spanned from i to i+n-1 and, then, the value of the corresponding array elements were added by 1. The site i was determined by a random number generated on computer. A contig is defined to be a stretch of array elements which hold a non-zero value. On the other hand, a gap is a continuum of array elements that hold zero value. Site redundancy is defined as the value of an array element at a particular site while redundancy, r; is defined to be Si A(i)/N. Coverage k is defined as (the total number of array elements which hold non-zero value)/ N.
The key parameters employed for composing the program are genome size(N), fragment size(n), redundancy(r), coverage(k), the number of contigs (j) , which are depicted in Figure 1. Genome DNA(s) which can be either single or plural (or fragmented) chromosomes were treated as a single linear molecule (called genome sequence) by imaginatively ligating those fragments into one (although this approximation introduces a minor problem caused by ligating chromosomes and the resulting end-vanishing, it can be negligible so far as n is substantially small (say, less than a hundredth of N). Each nucleotide of a genome sequence (i-th nucleotide) is represented by an array element (A(i): A has N-dimension) in computation. In the program(Yuki-com2), a random number, x (0<=x<=1) which has sufficiently large significant figures, is generated in order to determine the site for a sequence read (i.e., the starting point of the sequence (Ps) is calculated to be INT (N×x) (if Ps is greater than N-n, then the trial is abandoned in order to correct a kind of a terminal effect), where INT is a function used in Visual Basic. The elements of an array which correspond to the site sequenced, spanning from Ps to Ps+n-1, are uniformly added by 1. Thus, a contig (which means a contiguous sequence generated by ligation of determined sequences based on their overlap ) is a region of sequence which contains no "0"-value elements within. Coverage, k, and redundancy, r, are obtained from the equations below:


where d(A (i),0) is equal to 1 for i holding A (i) = 0, otherwise 0.
Simulations were repeated, usually, 1000-7000 times until the value of standard deviation of redundancy came into within 10 % of the mean value of redundancy. When f (=n/N) was small enough (such as less than 7.7×10-6) for law of large numbers to become effective, only two trials were sufficient to obtain a well-converged value of redundancy.
Therefore, this program has a nature of being usable for many phenomena which conform to Poisson distribution. This program( Yuki-com2) is available on request to the corresponding author (K.N.).
3 Results and Discussion
It is a priori clear that if the fraction value, f (=n/N), is the same for the different sets of parameters used for simulation, the results are identical due to the fractal nature of the problem concerned. In fact, if f was the same for various pairs of N and n, the results were expressed in a degenerated mode as in Figure 2 (including the data not shown). It is also evident that the smaller the value of f, the less the redundancy (r) at any coverage (k) is, which can be rationalized by the fact that when the degree of coverage become higher, the undetermined sequence portions (or gaps) turn smaller in size so that relatively long reads are apt to bear overlaps. This fact is intriguing because the average length of reads for human genome (3Gb in length) sequencing can be the same with the current one (400-500bp) applied to bacterial whole genome sequencing (around 1-4 Mb) without technical improvements to read longer and, nevertheless, with reduction in redundancy.
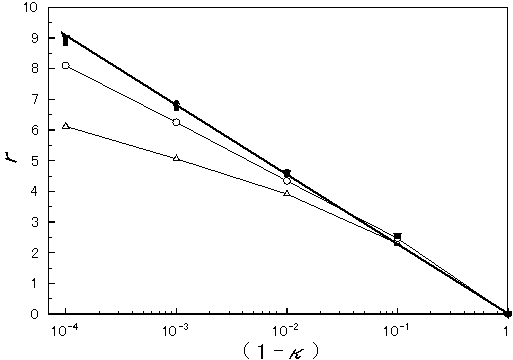
Figure 2. A plot of redundancy versus coverage. The abscissa is a processed value of coverage as 1-k. The ordinate is for redundancy. The symbols express the combination of parameters (the genome size (N) and the size of sequence read (n));  ((N=) 5.2×107 and (n=) 400),
((N=) 5.2×107 and (n=) 400),  (5.2×107, 100),
(5.2×107, 100),  (6.5×106, 400),
(6.5×106, 400),  (6.5×106, 100),
(6.5×106, 100), (6.5×106, 4), and
(6.5×106, 4), and  (6.5×105, 400), respectively. The thick line is a regression line for the relevant points, from which Eq.3 was obtained while the other thin lines are polygon lines.
(6.5×105, 400), respectively. The thick line is a regression line for the relevant points, from which Eq.3 was obtained while the other thin lines are polygon lines.
The upper limit dependence of r on 1-k shown in Figure 2 can be formulated as follows:

Equation 3 is useful for estimating the optimal value of k at which there must be a methodological shift from a random process of a shotgun to a directed process such as PCR relay [7], which can be called switching point. If we put the total cost for a whole genome sequencing as C and the costs of a shotgun process (which continues from the beginning to the switching point at the coverage of k) and a directed process for sequencing a unit length of fragment as cs and cd, respectively, C can be written as follows:

where l represents the average length of the sequences read (in this simulation, no size distribution assumed), and the subscripts, s and d, stand for 'shotgun' and 'directed', respectively. Using Eq.3 where r = rs, and subjecting the result to Taylor expansion, Eq.4 can be arranged as follows:
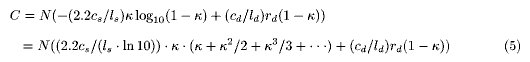
Therefore,
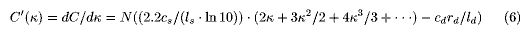
Since 0<=k<1 and all factors appearing in Eq.6 are positive, C' is monotonous. As C'(0)<0, C'(k) is equal to 0 at one and only value of k(kmin), where C has the minimum (Cmin) . kmin and Cmin can be numerically obtained using Eqs. 5 and 6. If cd / cs =3, ls ld(=400), and rd=2.2, kmin=0.84 ( where rs=1.8 from Eq.3). If cd / cs = 10 and the rest are the same with the above, kmin = 0.95 (rs=2.9). This means that depending on the cost of sequencing per nucleotide (c/l) for both the processes, shotgun and directed, we can optimize the total scheme (although we think the ongoing genome sequencing project must be more dynamically operated because of incessant technical improvements and thus the estimation performed here remains tentative). Eq.3 gives us an optimal (lowest at cost or shortest at term or so) solution for planning a shotgun-based/directed- process-complemented genome sequencing.
ld(=400), and rd=2.2, kmin=0.84 ( where rs=1.8 from Eq.3). If cd / cs = 10 and the rest are the same with the above, kmin = 0.95 (rs=2.9). This means that depending on the cost of sequencing per nucleotide (c/l) for both the processes, shotgun and directed, we can optimize the total scheme (although we think the ongoing genome sequencing project must be more dynamically operated because of incessant technical improvements and thus the estimation performed here remains tentative). Eq.3 gives us an optimal (lowest at cost or shortest at term or so) solution for planning a shotgun-based/directed- process-complemented genome sequencing.
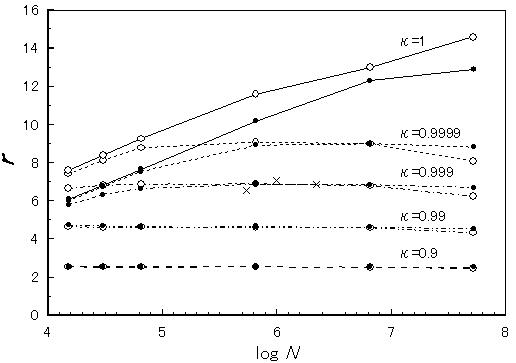
Figure 3. Dependence of redundancy on genome size (N) and coverage (k). The genome-size dependence of redundancy was examined by computer simulation at various coverage values: k=0.9, 0.99, 0.999, 0.9999, and 1 from bottom to top with either value of n =100 () or n=400 (), respectively. Some of the experimental values obtained from the actual genome sequencing projects (see Table 1) are plotted with a symbol(×).
Figure 2 also contains an important fact that the redundancy (r) of a shotgun process applied to human genome sequencing is, theoretically, at most 5 at the stage of 99% coverage (k=0.99) performed under possible conditions. In addition, there seems to be no substantial difference in efficiency between bacterial genome (Mb-sized) and human genome (Gb-sized) sequencing, which is more clearly shown in Figure 3. Rationally, due to its fractal nature, Gb-sized genome sequencing is approximately equivalent to 1000 times of Mb-sized one when a shotgun process is employed. Though computer works become complex and burdensome (e.g., to find a partially overlapping sequence out of n species of fragments, the amount of calculation becomes in proportion to n2, not n, therefore, at least in such an aspect, sequencing of a genome of m-fold length requires more than an m-fold amount of works), it is already well-known that the ratio of the computer work to the whole is rather small [6, 8].
Table 1 shows experimental scores of redundancy collected from the bacterial genome sequencing, mostly performed and published by Venter and his collaborators [8 - 14]. In average, genomes of Mb in size are sequenced at the redundancy of near 7. Some of those values are plotted in Figures 3, 4. If we assume that shotgun process was pursued up to 99 % coverage in those genome sequencing, those experimental values are larger by around 2 than those of simulation (Figure 3). The increment of the experimental values from those of simulation can be explained by the facts that a fraction of the length, Dl (>=log4N; which is, theoretically, the smallest number of a stretch of nucleotides in order to be unique in a genome of the size, N) is required to declare overlap and that there was, in reality, a size distribution in sequences read which is known to contribute to slightly increase redundancy [5] while, throughout this simulation, such effects were neglected. Moreover, in actual experiments there are inevitable errors and artifacts which greatly contribute increasing redundancy.
Table 1 Redundancy and the other scores in whole genome shotgun sequencing reported.
| # | Organism | Genome size (N) /bp | Average read length (n) /nts | Redundancy (r) | Number of contigs (j) | j/N ×105 | Ref.
|
|---|
| 1 | Haemophilus influenzae | 1,830,137 | 460 | 6* | - | - | [8]
|
| 2 | Mycoplasma genitalium | 580,070 | 435 | 6.5 | 39 | 6.7 | [7]
|
| 3 | Methanococcus jannaschii | 1.66×106 | 481 | 10.6* | - | - | [9]
|
| 4 | Helicobacter pylori | 1,667,867 | - | - | - | - | [10]
|
| 5 | Archaeoglobus fulgidus | 2,178,400 | 500* | 6.8 | 152 | 7 | [11]
|
| 6 | Borrelia burgdorferi | 910,725 +533,000 | 505 | 7.5 | - | - | [12]
|
| 7 | Treponema pallidum | 1,138,006 | 525 | ~7 | 130§ | >=11.4 | [13]
|
| 8 | Methanobacterium thermoautotrophium delta H | 1,751,377 | 361 | ~8.5* | - | - | [14]
|
* Calculated here based on the original data available. -, Not clear.
§ Clearly, single-sequence contigs are not included.
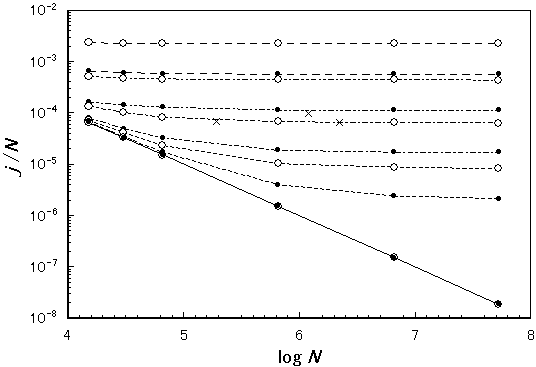
Figure 4. Dependence of the number of contigs per nucleotide (j/N) on genome size (N). The same symbols are used here as in Figure 3. Coverage values: k=0.9, 0.99, 0.999, 0.9999 and 1 from top to bottom.
It is far more significant than the absolute value of redundancy itself that there is almost no genome-size dependence in redundancy as shown in Figure 3. Thus, we can assume that the redundancy in human genome sequencing is not so different from that obtained from bacterial ones (r=7), which is a very favorable prediction for whole human genome shotgun sequencing. The number of contigs (j) per unit length is also rather independent on the genome size (Figure 4). This also supports the shotgun approach applied to human genome sequencing since the scale of end-closing process, which depends on the number of contigs (j), is proportional to the genome size. The experimental values for the number of contigs per nucleotide (j/N) indicate that 99% coverage was attained in those experiments, which is consistent with the assumption above introduced. (However, it should be noted that the number of contigs in our definition contains even the type of a single sequence while, in most of the works referred, single-sequences were omitted from contigs so that their values should have been counted less. The number of single-sequences is, unfortunately, not included in their papers but can be estimated to be less than j.) The number, j, enables us to estimate the average size of gaps, g, by the following equation:

Therefore, if N= 2000000, k= 0.95, j= 400 , then g = 250. If N= 2000000, k= 0.99, j=150 , then g = 134. Evidently, in the late stage of genome sequencing, the average size of gaps (g) is less than the size of a single sequence (n; ~400), which well agrees with the observation reported (g = less than 300bp) [8].
The most popular objection to the application of shotgun sequencing to human genome seems to come from the misunderstanding that end-closing process is or ought to be also a stochastic one. What is theoretically clear is that if the entire process of whole genome sequencing is subjected to a stochastic process, the increase of genome size (N) leads to a tremendous number of redundancy before the completion of sequencing as partly suggested in Figure 3 (rightward-increasing curves for k=1). (Plainly, if the probability for a genome to be completely sequenced within i-trials is p (0<= p<=1), then for a genome of the m-fold size to be completed within m×i-trials will be pm(<<1 if m =1000), which means an awfully rare event.) Therefore, especially in a vast genome, end-closing should be done by a directed process, of which an experimental amount is proportional to the size of the sequence to be read (i.e., N(1-k)). Since there is, actually, such a directed process as PCR relay [7, 16], shotgun sequencing of a vast genome is much feasible.
The redundancy for the whole processes, rw , is expressed as follows:

Thus, if the cost for a directed process (cd) is not so high in comparison with that for a shotgun process (cs) as in cd / cs= 3 (as discussed above as to Eq.6 ), then rw =1.9. On the other hand, if cd / cs= 10, then rw = 2.9. Thus, the redundancy value (rw) theoretically obtained is sufficiently small. The total costs for both the cases (assuming N=3.5×109) will amount to 2.2×107 cs (for rw = 1.9) and 3.4×107 cs (for rw = 2.9), although, in reality, the rw value for actual experiments must be roughly doubled as above discussed: C6×107 cs. This estimation offers us a very clear insight into the total cost because it demands only the cost for sequencing a single clone (~400bp); cs. Though it is difficult for us to accurately estimate the value of cs, if we could put it to be $ 10 (Currently, custom sequencing services are available at the cost of less than $ 20), the total cost will be around $ 600 million. This number is 3 times larger than that proposed by Venter et al. [1, 17].
The relatively high redundancy inherent to shotgun strategy is advantageous both for the accuracy of genome sequencing and for obtaining contigs. If two sequence reads are the same at a particular site and the error rate for each read is e (0<=e<=1), say 10-2, the final error rate at the site will be e2 ( =10-4). Likewise, if a particular site has the redundancy of three and more than 2 out of the 3 reads are the same, the error rate at the site will be less than 3(1-e)e2 (3×10-4). Therefore, the agreed error rate among the genome scientists for high quality genome sequencing (10-4) [3] can be attained, at least ideally, if more than 3 fold-coverage is secured throughout the whole genome sequence, relying on even low quality sequences of a one in 100 error rate or so. Another merit of high redundancy is to facilitate generating contigs. The larger the redundancy, the larger the overlap portion grows, which helps align sequences accurately and readily.
The other problems to be encountered in genome sequencing such as a problem of repeated sequences, which are not specific to whole genome shotgun sequencing but common to all strategies [4, 6], will be discussed elsewhere. This paper is intended to pursue a scientific and technological solution to large genome sequencing at large (not only to the present human genome sequencing) and not intended to side with a particular scientific policy.
4 Conclusions
The Monte-Carlo simulation enabled us to quantitatively estimate the redundancy in shotgun sequencing of vast genomes such as human genome. To evaluate the optimal coverage value (k), or the switching point, Equation 5 was introduced. The values thus obtained supported that whole genome shotgun sequencing can be a cost-saving strategy for human genome sequencing. If a directed process is not so costly in comparison with a shotgun process, we can reduce the redundancy to 3 by switching earlier. From the accuracy viewpoint, the redundancy of more than 3 was shown to be useful. Therefore, whole genome sequencing by a shotgun process up to 95-99% coverage and followed by a directed sequencing to completion will be, actually, the redundancy of ca. 7 and optimum.
This research was supported by Grant-in-Aid (#09272203) from the Ministry of Education, Science, Sports and Culture of Japan.
References
[ 1] Venter, J.C., Adams, M.D., Sutton, G.G., Kerlavage, A.R., Smith, H.O., and Hunkapiller, M., Science, 280, 1540-1542 (1998).
[ 2] Marshall, E., Science, 281, 1774-1775 (1998).
[ 3] Waterston, R. and Sulston, J.E., Science, 282, 53-54 (1998).
[ 4] Green, P., Genome Research, 7, 410-417 (1997).
[ 5] Lander, E.S. and Waterman, M.S., Genomics, 2, 231-239 (1988).
[ 6] Weber, L.J. and Myers, E.M., Genome Research, 7, 401-409 (1997).
[ 7] Nishigaki, K., Kinoshita, Y., Sakuma, Y., Nakabayashi, Y., Kyono, H ., and Husimi, Y., Sci. Eng. Rep. Saitama Univ., No.27, 43-52 (1993).
[ 8] Fraser, C.M. et al., Science, 270, 397-403 (1995).
[ 9] Fleischmann, R.D., et al., Science, 269, 496-512 (1995).
[ 10] Bult, C.J., et al., Science, 273, 1058-1073 (1996).
[ 11] Tomb, J-F., et al., Nature, 388, 539-547 (1997).
[ 12] Klenk, H-P., et al., Nature, 390, 364-370 (1997).
[ 13] Fraser, C.M., et al., Nature, 390, 580-586 (1997).
[ 14] Fraser, C.M., et al., Science, 281, 375-388 (1998).
[ 15] Smith, D.R., et al., J. Bacteriol., 179, 7135-7155 (1997).
[ 16] A paper dealing with this method will be soon published in a journal well circulated (one possibility is Genome Research).
[ 17] The tentative cost estimation ($ 600 million) fell on the amount which is nearly equivalent to that of the three-years-budget of National Human Genome Research Institute (NHGRI) [2] and less than a half of the original estimate (Watson, J.D., Science, 248, 44(1990)), although this estimation leaves much room to be reviewed.
Return